ARTICLE Photodissociation Dynamics of 2-Iodotoluene Investigated by Femtosecond Time-Resolved Mass Spectrometry†
2016-04-08ZhimingLiuYanmeiWangChunlongHuJinyouLongBingZhangStateKeyLaboratoryofMagneticResonanceandAtomicandMolecularPhysicsWuhanInstituteofPhysicsandMathematicsChineseAcademyofSciencesWuhan430071ChinaDatedReceivedonNovember
Zhi-ming Liu,Yan-mei Wang,Chun-long Hu,Jin-you Long,Bing Zhang∗State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics,Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences,Wuhan 430071,China(Dated:Received on November 15,2015;Accepted on January 14,2016)
ARTICLE Photodissociation Dynamics of 2-Iodotoluene Investigated by Femtosecond Time-Resolved Mass Spectrometry†
Zhi-ming Liu,Yan-mei Wang,Chun-long Hu,Jin-you Long,Bing Zhang∗
State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics,Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences,Wuhan 430071,China
(Dated:Received on November 15,2015;Accepted on January 14,2016)
The photodissociation dynamics of 2-iodotoluene following excitation at 266 nm have been investigated employing femtosecond time-resolved mass spectrometry.The photofragments are detected by multiphoton ionization using an intense laser fi eld centered at 800 nm.A dissociation time of 380±50 fs was measured from the rising time of the co-fragments of toluene radical(C7H7)and iodine atom(I),which is attributed to the averaged time needed for the C−I bond breaking for the simultaneously excited nσ∗and ππ∗states by 266 nm pump light.In addition,a probe light centered at 298.23 nm corresponding to resonance wavelength of ground-state iodine atom is used to selectively ionize ground-state iodine atoms generated from the dissociation of initially populated nσ∗and ππ∗states.And a rise time of 400±50 fs is extracted from the fi tting of time-dependent I+transient,which is in agreement with the dissociation time obtained by multiphoton ionization with 800 nm,suggesting that the main dissociative products are ground-state iodine atoms.
Key words:2-Iodotoluene,Photodissociation,Dissociation time,Femtosecond timeresolved mass spectrometry
†Part of the special issue for“the Chinese Chemical Society’s 14th National Chemical Dynamics Symposium”.
∗Author to whom correspondence should be addressed.E-mail: bzhang@wipm.ac.cn,Tel.:+86-27-87197441,FAX:+86-27-87198491
I.INTRODUCTION
The photodissociation dynamics of halogenated organic compounds has attracted historically great attention,not only due to its crucial role in highly detailed fundamental studies but also its harmfulness to environment[1−17].The principal goal of photodissociation studies is to obtain the clearest picture of the molecular dynamics in the excited state as the molecule leaves the Franck-Condon region such as what transient states to traverse,where the fragments are formed,what the lifetime of the upper state is and which bonds break and so on.Many experimental techniques have been developed to investigate photodissociation reaction process,for instance photofragment translation spectroscopy[4−6], velocity map imaging[7,8]and ultrafast time-resolved time-of- fl ight mass spectrometry[9−15],which are coupled with high level ab initio calculations[15−17]to provide a clear picture of the fragmentation mechanisms of molecules.
The UV absorption spectra of aryl iodine are dominated by two contributions:one is the nσ∗states resulting from the promotion of a nonbonding electron from the iodine atom valence shell to an antibonding σ∗localized along the C−I bond,which leads to a rapid direct dissociation,and the other is due to the absorption to the bound ππ∗states of the benzene ring which has predissociation character for the coupling with repulsive nσ∗states.As a result of the overlap of these two di ff erent type states in the same energy region,the dynamics cannot be directly obtained from the absorption spectrum.Femtosecond time-resolved mass spectrometry coupled with state-selective resonance enhanced multiphoton ionization has been emerging as a powerful tool for investigating photodissciation dynamics process.It has two major advantages:the fi rst one is mass selectivity,which allows for the separate study of the reactant and fragment dynamics,the second one is the ability of selectively monitoring all fragments of di ff erent masses or same masses in di ff erent fi nal state as well as the parent ion simultaneously.
2-Iodotoluene,which is formed by substitution of an iodine atom and a methyl group for two neighboring hydrogen atoms of the benzene,is a very interesting system for study.Using state-selective onedimensional photofragment translational spectroscopy, El-Sayed et al.investigated the photodissociation dynamics of 2-iodotoluene upon excitation at 266 nm[6]. The spatial and velocity distributions of ground-state iodine atom I and excited state iodine atom I∗were determined.Two distinct ground-state iodine atoms velocity distributions were observed.One is a high velocity,narrow distribution with high anisotropy,whichis attributed to direct dissociation from the repulsive nσ∗state.The other one is a low velocity,broader distribution appearing lower anisotropy,which is due to the predissociation from the bound ππ∗state.The dissociation times for these two channels were estimated by calculating time-dependence of anisotropy parameter β.Fang et al.have calculated the potential energy curves for the ground and low-lying excited states of 2-iodotoluene along the assumed photolysis reaction coordinates and elucidated the dissociation mechanism and channels following excitation at 266 nm[17].
Based on the previous studies on aryl halides,in this work,we identify the photodissociation dynamics of 2-iodotoluene and the processes that participate in the relaxation of the molecule after being initiated by excitation at 266 nm.Especially,the determination of time scales for the dissociation channels is the focus on. Although previous experiment has estimated the dissociation times through measurements of the anisotropy [6],the accuracy is low especially when reaction time is more or comparable to the average rotation time of the parent molecule.Femtosecond time-resolved mass spectrometry enables one to directly measure the dissociation time.To the best of our knowledge,this is the fi rst time-resolved study on the photoinduced C−I bond breaking of 2-iodotoluene at 266 nm.
II.EXPERIMENTAL SETUP
The details of the experimental setup are described elsewhere[18].Brie fl y,it consists of a molecular beam machine coupled to a linear time of fl ight mass spectrometer and a 1 kHz-4 mJ/pulse Ti:Sapphire regenerative-ampli fi ed laser system(Coherent Inc.),delivering pulses with a central wavelength of~800 nm and a Fourier-transform-limited full width at halfmaximum(FWHM)duration of~100 fs.One part of the output light was used to produce the pump pulse at 266 nm by mixing the fundamental(800 nm)and the second harmonic beam(400 nm)in a 0.2 mm thick BBO crystal.One part was applied to pump an optical parametric ampli fi er(OPA,Coherent Inc.TOPAS-C) to generate probe wavelength centered at 298.23 nm, which is used for the resonance-enhanced multiphoton ionization(REMPI)probing of ground-state I atoms. Another part was used as probe light to track the relaxation processes of 2-iodotoluene.The energy intensity for pump light is 0.5µJ/pulse which keeps no ion signal occurring with it alone.For the probe light,the typical energy is 10 and 60µJ/pulse for the 298.23 and 800 nm respectively.The probe beam was temporally delayed relative to the pump beam by a computer-controlled linear translation stage(PI,M-126.CG1).The two laser beams were focused with fused silica lens of f=400 mm respectively and introduced into the vacuum chamber collinearly through a dichroic mirror.
The employed apparatus[19]is similar to that designed by Eppink and Parker[20].It consists of a molecular-beam source chamber and an ionization- fl ight detection chamber.The detection chamber was kept below 0.5µPa with the molecular beam on.2-Iodotoluene (99.9%purity)seeded in He was expanded into the source chamber with a stagnation pressure of 2 atm through a pulsed nozzle(General Valve,with a 0.5 mm ori fi ce)with the repetition rate of 10 Hz.The supersonic molecular beam is collimated by a conical skimmer and intersects perpendicularly with the two laser beams in a two-stage ion lens region.Photoion is extracted into a 36 cm fi eld-free region,which is doubly shielded against stray magnetic fi elds byµ-metal tube.At the end of the time-of- fl ight tube,the ions strike a twostage microchannel plate detector backed by a phosphor screen.The emission from the phosphor screen is monitored by a photomultiplier connected to a 1 GS/s digital oscilloscope(Tektronix Inc.,TDS2012B)USB interfaced with a computer.The LabView software was used to track the parent ion and fragments signal as function of pump-probe delay time simultaneously.
III.RESULTS AND DISCUSSION
With the help of detailed theoretical calculation[17] and experimental investigation[6]on 2-iodotoluene,it is easy to determine that upon excitation at 266 nm,the dominant excited states are the ππ∗state with a bound character and the nσ∗state with a repulsive character along the C−I stretching coordinate,which leads to the production of ground-state iodine atoms.So,the parallel relaxation processes for those two excited states are monitored by probe pulse which ionized the excited molecules.
Figure 1(a)displays the time-of- fl ight mass spectra obtained with the pump pulse at 266 nm alone,the probe pulse at 800 nm alone and in pump-probe con fi guration at∆t=0(time-overlap).The power of the probe light is~26µJ/pulse.As observed in the mass spectra, there are many fragment ions generated other than parent ion C7H7I+,especially for toluene ion C7H7+which is the dominant signal in the mass spectra,indicating that the parent ion will to a large extent undergo photoinduced fragmentation[21,22],which is similar to the femtosecond pump-probe investigation on iodobenzene [10].Figure 1(b)shows the measured time transients of the total signals of,following excitation at 266 nm and probe with 800 nm(26µJ/pulse). The polarizations of pump and probe beam are parallel with each other.The decay time pro fi le for parent ioncan be best fi tted by one exponential with decay time constant τdof 92±10 and 129±15 fs respectively convoluted with a Gaussian that describes the pump-probe cross correlation.
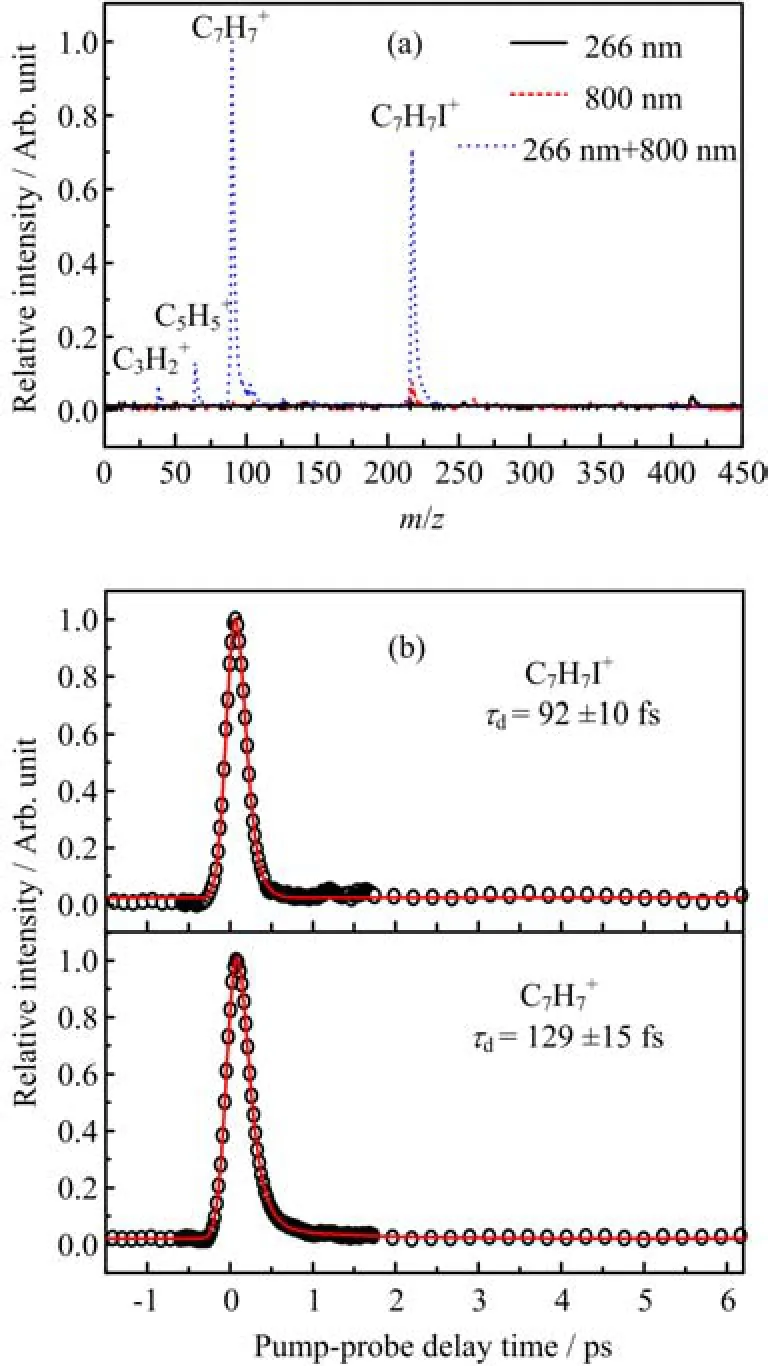
FIG.1(a)One color and two color(at time overlap)mass spectra of 2-iodotoluene at 266 and 800 nm.The typical energy was 0.5 and 26µJ/pulse for the 266 nm pump and 800 nm probe pulse respectively.(b)Time-resolved C7H7I+and C7H7+transients recorded under the same conditions as(a),the circles represent experimental data and the solid lines are fi tting results.
It is not likely that those fragment ions shown in Fig.1(a)are produced from the ionization of the corresponding neutral radicals,which is generated from dissociation of neutral molecules or fragmentation of molecular ion,due to their high ionization potential under our low probe light intensity.For instance,ionization of neutral toluene radical would require absorption at least six probe photons(800 nm)since the ionization potential of the toluene radical is expected to be only slightly lower than the ionization potential of the phenyl radical,which is 9.13 eV[23,24].If the neutral radicals can be detected,the time-pro fi le of C7H7+should have a stable channel at longer pump-probe delay time, which is not observed on the fragment ion in Fig.1(b), indicating that the neutral radical C7H7generated from dissociation in electronically excited states induced by the pump pulse is not ionized by the probe pulse 800 nm with intensity of 26µJ/pulse.So,it is expected that all fragment ions observed here are originated from dissociative ionization of parent molecules.Since no ion signals were generated with pump pulse alone,the timedependent fragment ions signal should also re fl ect the neutral excited states dynamics of 2-iodotoluene.
The repulsive nσ∗and the bound ππ∗states can be excited simultaneously.So,it is expected that two decay components should be observed on the time pro fi le of parent ion or fragment ions.Such kinetics is observed on iodobenzene molecule[10].Unfortunately,only one decay component is observed for C7H7I+and C7H7+transients shown in Fig.1(b)and we tend to attribute this component to the decay dynamics of the initially populated nσ∗state.There are two possible reasons for not observing the contribution from ππ∗state:one is that the absorption cross-section of the ππ∗state is much lower than the nσ∗state and indeed the absorption coe ffi cient of the nσ∗state is an order larger than that of the ππ∗state according to the theoretical calculation[7];another one is that the ionization crosssection for the ππ∗state is probable low with 800 nm as probe light.Thus,the ionization signal from the ππ∗state would be faint,which is likely to be suppressed by strong ionization signal from the nσ∗state.
Now we turn to discuss the observed lifetime τd, which is assigned to the decay dynamics of the initial populated nσ∗state as mentioned above.Indeed,a very fast relaxation will occur when excited to this state due to its repulsive character.Using state-selective photofragment translational spectroscopy Freitas and coworkers[6]studied the photodissociation dynamics of 2-iodotoluene upon excitation by 266 nm and observed a sharp high velocity distribution of ground-state iodine atoms,which was assigned to a direct dissociation occurring on the nσ∗state.And they estimated the dissociation time(0.51 ps)of C−I bond on this state by calculating β variation as a function of time.According to the calculations[17],it has big possibility for the molecules to stay on this state during decay processes.Therefore,the observed lifetime for the decay of nσ∗state should be shorter than the dissociation time which is associated with the time needed from the Franck-Condon region to the production of freedom iodine atoms.Thus,the fi tted τdis reasonable and not contradicted with the estimated dissociation time of 0.51 ps in Freitas’work.It is persuasive to assign the lifetime constant τdto the decay dynamics on the initially excited nσ∗state.
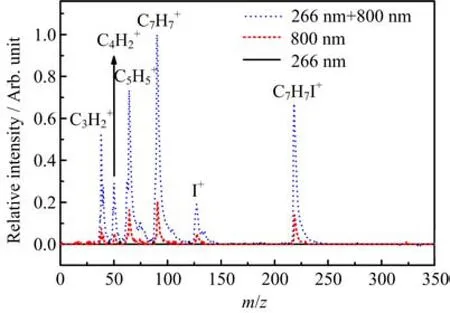
FIG.2 The mass spectra of 2-iodotoluene obtained with 266 nm pump light alone and 800 nm probe light alone and at time overlap between pump and probe light.The typical energy was 0.5 and 60µJ/pulse for the 266 nm pump and 800 nm probe pulse respectively.
In order to get more insight to the dissociation dynamics of the excited states induced by 266 nm,we increase the power of probe pulse to try to detect the dissociative products.It is an ideal tool to detect molecular species with easily achieved multiphoton ionization using femtosecond lasers[25].By increasing the power of the probe beam from 26µJ/pulse to 60µJ/pulse, a time-of- fl ight mass spectrum recorded at pump-probe delay time zero is shown in Fig.2.More visible fragment ions signal intensity is observed compared with the mass spectrum obtained with probe intensity 26µJ/pulse. To follow the transient dynamics for these fragment ions,the signal intensities against the pump-probe delay time for these peaks were acquired,which is shown in Fig.3.The temporal behavior of all fragment ions can be well fi tted to one fast decay component and one rise component,convoluted with a Gaussian describing the pump-probe cross correlation.All decay and rise times measured for all cation transients are summarized in Table I.Those two components correspond to two di ff erent channels for the production of the fragment ions.For the fast decay time constant τd,it is reasonable to attribute this component to the dissociation of the parent ions after the pump-probe ionization, since it has similar trend to the parention time pro fi le shown in Fig.3.For the second rising component τr,it is a constant component without decay.Furthermore,this component depends greatly on the power of the probe beam.Thus this component is attributed to the ionization of the neutral fragments generated by the pump pulse.As mentioned above,the dissociative products are toluene radical C7H7and iodine atom I induced by the pump light.So,it is easy to assign the rise component fortransients to the ionization of neutral C7H7and I.It is very interesting to observe that thetransients also show the rise component.By closely inspecting the time pro fi le of these transients,it is discovered that the time constants τrare in agreement with that forThus,it is likely that the rise components intransients are from the dissociative ionization of neutral radical C7H7.Indeed,there are no dissociative channels from the parent molecules to product neutral radical C5H4,C4H2and C3H2after being excited by 266 nm,which further evidence our conclusion.So,the τrcomponents in these fragment ions transients re fl ect the same dynamics as in. It is worth noting that the rise time constant τrre fl ects the averaged time needed for all dissociative channels initialed by 266 nm,leading to generation of neutral radical C7H7and iodine atom I.

FIG.3 Time-resolved cation transients recorded under the same conditions as Fig.2.The circles represent experimental data and the solid lines are fi tting results.All transients are normalized to their maxima value.

TABLE I The time constants extracted from the fi ts of all cation transients shown in Fig.3.
It is interesting that the decay times increase as the mass of the fragment ions decreases.As mentioned above,the decay components in all fragment ions transients are from the fragmentation after pump-probe ionization of parent molecule.These observations are similar to the ones obtained,for instance,on tetrathiafulvalene and Cr(CO)6probed with an intense probe pulse[26,27].Here as well,the degree of fragmentation is weaker at small pump-probe delays and becomes stronger at long pump-probe delays.As explained,when the molecule relaxes,the electronic energy is converted into vibrational energy,which remains in the ion upon ionization.Dissociative ionization is sensitive to vibrational relaxation since the bonds in the ion are generally weaker than those of the neutral molecules. Therefore,upon ionization,the internal energy of the ion will be more and more important and fragmentation to the smaller species will take place once intramolecular vibrational relaxation becomes e ff ective[26].
To gain more information on the photodissociation dynamics of 2-iodotoluene upon excitation by 266 nm, the probe light at 298.23 nm,corresponding to the ground-state iodine atom resonance wavelength[28],is used to track the appearance of ground state iodine atoms product generated by cleavage of C−I bond in this molecule.There are no excited state I atom resonance wavelengths within the bandwidth of the probe light(450 cm−1),which enables us to probe the ground state iodine atoms only.Figure 4 displays the I+transient obtained following excitation at 266 nm and probe with 298.23 nm.It can be fi tted with a single rising exponential convoluted with the Gaussian pumpprobe cross correlation,yielding a rising time constant of 400±50 fs,which re fl ects the average time for all dissociative channels leading to ground state I atoms. Two distinct ground-state I atom spatial and velocity distributions were observed by El-Sayed’s group[6] using nanosecond lasers.One is a high velocity,narrow distribution that exhibits a high anisotropy,which is assigned to the direct dissociation from nσ∗state. The other one is a low velocity,broader distribution accompanied by a lower averaged anisotropy,which is attributed to the predissociation dynamics of ππ∗state. And they speculated that the dominant products of ground-state I atoms resulted from direct dissociation process.The dissociation time of this process is estimated to be 0.51 ps.The value 400±50 fs extracted from the time transients of ground-state I atom in our experiment is in agreement with the estimated value 0.51 ps[6].Therefore,it is reasonable to conclude that the measured rise time 400±50 fs dominantly re fl ects the dissociation time needed for C−I fi ssion on the nσ∗state populated by 266 nm,though we cannot rule out the contribution from the predissociation dynamics of initially excited ππ∗state.
IV.CONCLUSION
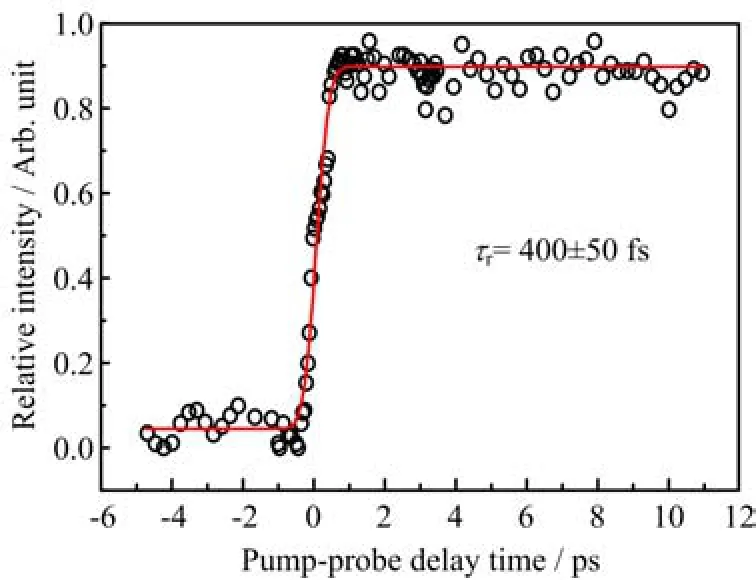
FIG.4 The I+transient obtained following excitation at 266 nm and probing at 298.23 nm corresponding to the resonance wavelength of ground state iodine atom.The circles are experimental data and the solid line is the fi tting result.
The ultrafast relaxation of 2-iodotoluene has been studied after excitation at 266 nm with the goal of determining the time scales of the dissociation channels.In this excitation wavelength,a repulsive nσ∗state and a bound ππ∗state are populated simultaneously.The obtained time-pro fi le of parent ion(C7H7I+)with 800 nm as probe light(26 or 60µJ/pulse)can be best fi tted by one decay exponential convoluted with a Gaussian describing the pump-probe cross correlation.However, a decay component and a rise component are needed to fi t the temporal behavior of all fragment ionsobtained with strong intensity of 800 nm probe light(60µJ/pulse).The decay time constant τd(105−245 fs)for all ions re fl ects the decay dynamics of the initially populated repulsive nσ∗state.While the rise time constant τr(380−405 fs)is attributed to the averaged dissociation time for all the dissociation channels induced by pump light 266 nm.In addition,we selectively tracked the dissociation channels leading to ground-state iodine atoms using groundstate iodine atom resonance wavelength of 298.23 nm as probe light.The measured appearance time 400±50 fs for I+transient is in agreement with the dissociation time obtained with multiphoton ionization with 800 nm as probe light,which indicates that the main products of dissociation induced by 266 nm are ground-state iodine atoms.
V.ACKNOWLEDGMENTS
This work was supported by the National BasicResearchProgramofChina(973Program) (No.2013CB922200)and the National Natural Science Foundation of China(No.91121006,No.21273274, No.21173256,and No.21303255).
[1]R.S.Mulliken,J.Chem.Phys.8,382(1940).
[2]R.K.Sparks,K.L.Shobatake,R.Carlson,and Y.T. Lee,J.Chem.Phys.75,3838(1981).
[3]J.L.Knee,L.R.Khundkar,and A.H.Zewail,J.Chem. Phys.83,1996(1985).
[4]H.J.Hwang and M.A.El-Sayed,J.Chem.Phys.94, 4877(1991).
[5]H.J.Hwang and M.A.El-Sayed,J.Phys.Chem.6, 8725(1992).
[6]J.E.Freitas,H.J.Hwang,and M.A.El-Sayed,J.Phys. Chem.98,3322(1994).
[7]X.B.Zhang,Z.R.Wei,Y.Tang,T.J.Chao,B.Zhang, and K.C.Lin,ChemPhysChem.9,1130(2008).
[8]Y.Tang,W.B.Lee,B.Zhang,and K.C.Lin,J.Phys. Chem.A 112,1421(2008).
[9]P.Y.Cheng,D.Zhong,and A.H.Zewail,Chem.Phys. Lett.237,399(1995).
[10]M.Kadi,J.Davidsson,A.N.Tarnovsky,M.Rasmusson,and E.˚Akesson,Chem.Phys.Lett.350,93(2001).
[11]M.Kadi and J.Davidsson,J.Chem.Phys.Lett.378, 172(2003).
[12]M.Kadi,E.Ivasson,and J.Davidsson,Chem.Phys. Lett.384,35(2004).
[13]R.Montero,A.P.Conde,A.Longarte,F.Casta˜no,M. E.Corrales,R.de Nalda,and L.Ba˜nares,Phys.Chem. Chem.Phys.12,7988(2010).
[14]G.Gitzinger,M.E.Corrales,V.Loriot,G.A.Amaral,R.de Nalda,and L.Ba˜nares,J.Chem.Phys.132, 24313(2010).
[15]O.A.Brog,Y.J.Liu,P.Persson,S.Lunell,D.Karlsson,M.Kadi,and M.Davidsson,J.Phys.Chem.A 110,7045(2006).
[16]Y.J.Liu,P.Persson,H.O.Karlsson,and S.Lunell,J. Chem.Phys.120,6502(2004).
[17]Y.J.Liu,Y.C.Tian,and W.H.Fang,J.Chem.Phys. 132,014306(2010).
[18]Y.Z.Liu,B.F.Tang,H.Shen,S.Zhang,and B.Zhang, Opt.Express.18,5791(2010).
[19]C.C.Qin,Y.Z.Liu,S.Zhang,Y.M.Wang,Y.Tang, and B.Zhang,Phys.Rev.A 83,033423(2011).
[20]A.T.J.B.Eppink and D.H.Parker,Rev.Sci.Instrum. 68,3477(1997).
[21]J.C.Lorquet and B.Leyh,Org.Mass Spectrum.28, 1225(1993).
[22]B.D.Koplitz and J.K.McVey,J.Chem.Phys.81, 4963(1984).
[23]C.F.Logan,J.C.Ma,and P.Chen,J.Am.Chem.Soc. 116,2137(1994).
[24]A.Nicolaides,D.M.Smith,F.Jensen,and L.Radom, J.Am.Chem.Soc.119,8083(1997).
[25]J.Peng,N.Puskas,P.B.Corkum,D.M.Rayner,and A.V.Loboda,Anal.Chem.84,5633(2012).
[26]D.Staedter,N.Thir´e,L.Polizzi,Y.Mairesse,P.Mayer, and V.Blanchet,J.Chem.Phys.142,194306(2015).
[27]S.A.Trushin,W.Fuss,W.E.Schmid,and K.L. Kompa,J.Phys.Chem.A 102,4129(1998).
[28]Y.J.Jung,Y.S.Kim,W.K.Kang,and K.H.Jung, J.Chem.Phys.107,7187(1997).
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- ARTICLE E ffi cient Separation of Ar and Kr from Environmental Samples for Trace Radioactive Noble Gas Detection†
- REVIEW Polarization Dependent Time-Resolved Infrared Spectroscopy and Its Applications†
- ARTICLE Reactions of Group V Metal Atoms with Hydrogen Sul fi de:Argon Matrix Infrared Spectra and Theoretical Calculations†
- ARTICLE Structural Dynamics of Phenyl Azide in Light-Absorbing Excited States: Resonance Raman and Quantum Mechanical Calculation Study†
- ARTICLE Structural and Infrared Spectroscopic Study on Solvation of Acetylene by Protonated Water Molecules†
- ARTICLE Excited-State Proton Transfer and Decay in Hydrogen-Bonded Oxazole System:MS-CASPT2//CASSCF Study†