ARTICLE E ffi cient Separation of Ar and Kr from Environmental Samples for Trace Radioactive Noble Gas Detection†
2016-04-08LeyiTuGuominYngXingyngZhngShuimingHuHefeiNtionlLbortoryforPhysiclSciencestheMicroscleUniversityofSciencendTechnologyofChinHefei230026ChinbInstituteofHydrogeologyndEnvironmentlGeologyChineseAcdemyofGeologiclSciences
Le-yi Tu,Guo-min Yng,Xing-yng Zhng,b,Shui-ming Hu∗.Hefei Ntionl Lbortory for Physicl Sciences t the Microscle,University of Science nd Technology of Chin,Hefei 230026,Chinb.Institute of Hydrogeology nd Environmentl Geology,Chinese Acdemy of Geologicl Sciences, Zhengding 050803,Chin(Dted:Received on October 9,2015;Accepted on December 24,2015)
ARTICLE E ffi cient Separation of Ar and Kr from Environmental Samples for Trace Radioactive Noble Gas Detection†
Le-yi Tua,Guo-min Yanga,Xiang-yang Zhanga,b,Shui-ming Hua∗
a.Hefei National Laboratory for Physical Sciences at the Microscale,University of Science and Technology of China,Hefei 230026,China
b.Institute of Hydrogeology and Environmental Geology,Chinese Academy of Geological Sciences, Zhengding 050803,China
(Dated:Received on October 9,2015;Accepted on December 24,2015)
Radioactive noble-gas isotopes,85Kr(half-life t1/2=10.8 y),39Ar(t1/2=269 y),and81Kr (t1/2=229,000 y),are ideal tracers and can be detected by atom trap trace analysis(ATTA), a laser-based technique,from environmental samples like air and groundwater.Prior to ATTA measurements,it is necessary to e ffi ciently extract krypton and argon gases from samples.Using a combination of cryogenic distillation,titanium chemical reaction and gas chromatography,we demonstrate that we can recover both krypton and argon gases from 1−10 L“air-like”samples with yields in excess of 90%and 98%,respectively,which meet well the requirements for ATTA measurements.A group of testing samples are analyzed to verify the performance of the system,including two groundwater samples obtained from north China plain.
Key words:Atom trap trace analysis,Gas chromatography,Radioactive noble gas
†Part of the special issue for“the Chinese Chemical Society’s 14th National Chemical Dynamics Symposium”.
∗Author to whom correspondence should be addressed.E-mail: smhu@ustc.edu.cn
I.INTRODUCTION
Owing to the unique properties of noble gases,three radioactive isotopes,85Kr(half-life t1/2=10.8 y),39Ar (t1/2=269 y)and81Kr(t1/2=229 ky),are homogeneously distributed in the atmosphere,and have simple mixing and transportation mechanisms in the environment.They are considered as ideal tracers in various studies,including groundwater dating,ocean ventilation,and nuclear safety.85Kr is a fi ssion product emitted to the northern hemisphere since the nuclear age[1].85Kr can be used to monitor anthropic nuclear activities and to calibrate atmospheric transport models[2,3].It can also be used as a tracer for dating young groundwater with an age range of 2−50 y[4−8].81Kr is a cosmogenic nuclide,and human nuclear activities have no detectable e ff ect on the abundance of81Kr.These outstanding characteristics make81Kr a desired tracer for dating old groundwater[9−12]and ices[13]on the time scale of 50−1000 ky.39Ar in the atmosphere is also produced by cosmic-ray,which is particularly interested for studies of deep ocean mixing and circulation on a time scale of 50−1000 y, fi lling a time window inaccessible by other radioactive tracers[14−17].
The concentration of krypton in the earth’s atmosphere is 1.14 ppm(part per million)by volume[18]. The isotopic abundances of85Kr and81Kr have been determined to be 2.2×10−11and(5.2±0.6)×10−13,respectively[2,19−21].Argon constitutes 0.934%of the atmosphere by volume,larger than krypton by four orders of magnitude,but the isotopic abundance of39Ar is only 8×10−16.85Kr and39Ar can be analyzed by lowlevel counting(LLC)of the decay.The minimal sample size of LLC analysis of85Kr is about 10µL krypton (STP,standard temperature and pressure),and several hundred milliliters argon(STP)for39Ar.The later one corresponds to a groundwater sample size of several tons[21].Due to much longer half-life time of81Kr, it is impractical to analyze81Kr with LLC.Accelerator mass spectrometry(AMS)has been successfully applied for81Kr-dating,but the sample size was huge:about 500µL krypton gas recovered from 16 ton groundwater [10].Atom trap trace analysis(ATTA)[22]is a laserbased technique,utilizing a magneto-optical trap to selectively capture and count atoms.The minimum krypton sample size for85Kr/81Kr detection with ATTA has been reduced to a few microliter[23−25].ATTA analysis of39Ar also becomes feasible[26,27].It has been concluded[28]that ATTA is currently the most practical method of dating environmental samples using radioactive krypton and argon isotopes.
One liter of modern groundwater at 10◦C contains about 5800085Kr atoms,130081Kr atoms,and 850039Ar atoms[29,30].Currently,ATTA measurement of radio-krypton needs a typical groundwater sample size of about 100 L.Prior to the ATTA measurement,it is necessary to extract noble gases(mostly argon andkrypton)from groundwater samples,and it can be accomplished in two steps: fi rst to extract the solved gas from groundwater,and then to separate krypton/argon from the“air-like”gas sample.Taking into account the complicated transfer and mixing of groundwater,analysis with multiple tracers is preferred in groundwater dating.Therefore,it is desired to separate and recover both argon and krypton from di ff erent gas samples with high yields to prevent any possible isotopic fractionation.
There have been several reports on the systems of Kr separation from“air-like”gases[5,10,25,31−33]. A method based on frozen charcoal trap and gas chromatography[5,32]has been applied for several liters of gas samples extracted from groundwater.A special krypton puri fi cation system for more than 100 L of bulk gas was reported by using several gas chromatographic steps,which has been applied in81Kr dating with AMS [10].Systems for recovering krypton from gases with a volume in the range of 1−100 liters have also been built based on cryogenic distillation,gas chromatography,and titanium reactions[25,33],and they have been successfully applied in radio-krypton dating measurements using ATTA.Here we report on a new system developed to recover both krypton and argon for ATTA measurements using 1−10 L gas extracted from groundwater samples.Using a combined process of cryogenic distillation,gas chromatography and titanium reaction, yields in excess of 90%and 99%have been achieved for krypton and argon,respectively.As a demonstration, abundances of85Kr and81Kr in several environmental samples have been determined by the ATTA instrument in Hefei(China).
II.EXPERIMENTS
A.Sampling in the fi eld:extract gases from groundwater
The con fi guration of the sampling system for fi eld sampling is shown in Fig.1.A membrane contactor (Liquicel,4×13,type X40)is used to extract gases from groundwater.The hydrophobic hollow- fi bre membrane contactor can e ffi ciently separate gases from liquid[34],and has been widely used in di ff erent applications[35−38].High e ffi ciency and simple structure make it very suitable to be used in fi eld.Groundwater sample fi rst passes through two fi ne fi lters to remove particles in the sample,then is introduced into the membrane contactor with a fl ow rate of 5−20 L/min monitored by a water fl ow meter.The contactor allows gases to di ff use from the water into gas- fi lled contactor pores which contact with gas line directly.The gas line is fi rst evacuated by a diaphragm pump and is further purged by the gas extracted from groundwater.When the size of the residual air is believed to be negligible, the extracted gas will be pumped into a sample cylinder by the diaphragm pump.Under a water fl ow rate of 10 L/min,about 5 L aqueous gas can be collected in about 0.5 h,with an extraction e ffi ciency of about 90% for Ar and O2and 70%for Kr.The exhaust end of the diaphragm pump connects with a sample cylinder,and the pressure of the fi nal collected gas is limited to be about 1.2 bar.More gas can be collected in the same cylinder if a compressor pump is used,but it considerably increases the weight of the system and consumes more power in the fi eld.

FIG.1 Schematic of the system for groundwater degassing. Abbreviations:W1 and W2:water valve,V1 and V2:threeway valve,P:pressure gauge.
Gas samples extracted from groundwater are contained in cylinders in the fi eld.Noble gases in the samples,mainly argon and krypton,will be extracted in the laboratories using cryogenic distillation and hightemperature Ti-reaction,followed by gas chromatographic separation.A schematic of the puri fi cation system is shown in Fig.2.
B.Cryogenic distillation and high-temperature Ti-reaction
Water vapor and carbon dioxide are fi rst removed by a molecular sieve 5˚A trap(MS 5A),then the gas sample is introduced into a liquid-N2(77 K)cooled charcoal trap(trap 1,200 cm3volume,4 g charcoal of 16−32 mesh).A vacuum compressor is used and typically it takes about 30 min to condense more than 95% of the gas sample into trap 1.The vapor above the condensed sample in trap 1 fl ows into a quartz tube with a fl ow rate of about 50 mL/min,which is constrained by a mass fl ow controller.The quartz tube(“burner”) is 34 mm in diameter,70 cm long,and O-ring sealed at both ends.The tube has been fi lled with about 200 g titanium sponge,and slowly heated to 1000◦C by a furnace.At temperature of 1000◦C,titanium reacts with gaseous O2and N2to form titanium oxides and titanium nitrides,respectively.Titanium also consumes other chemically active gases,including CH4at such high temperature.Consequently,residual gases in the burner are mostly noble gases,together with little amounts of N2and CH4.The fl ow rate of 50 mL/min isselected to keep a mild reaction rate to avoid overheating the burner.During the process,the pressure in the quartz tube is monitored with a gauge(MKS Baratron 627B,relative accuracy of 0.12%).Because krypton has a lower vapor pressure in liquid-N2cooled charcoal trap, krypton is kept condensed in trap 1,while most Ar,O2, and N2gases are released to the burner.

FIG.2 Schematic of the system to separate krypton and argon from air-like samples.MS 5A:molecular sieve 5˚A trap. Trap:frozen trap with activated charcoal.Furnace:furnace for titanium-reaction.Getter:titanium getter pump.GC:gas chromatography.
When the argon gas accumulates in the burner,the gas pressure gets higher and prevents the gas fl ow from trap 1 to the burner.At this point,we turn o ff the gas fl ow into the burner.The gas pressure in the burner will decrease since the Ti-reaction continues.It takes about 20 minutes to reach an equilibrium,which is illustrated in Fig.3(a).The residual gas(mostly argon)will be collected with another liquid-N2cooled charcoal trap (trap 2).Subsequently,we can turn on the gas fl ow from trap 1 to the burner again and restart the distillation. The procedure above can be repeated until most gas in Trap 1 is transferred,which is evidenced by a sudden drop of fl ow rate from trap 1 to the burner.Usually two iterations are needed for a sample with an original size of 10 L.We have investigated the composition of the gas fl ow during the process using gas chromatographic analysis,which is shown in Fig.3(b).At the beginning, the main composition is N2.Later when N2depletes, O2and Ar become dominant in the fl ow.Finally,the fl ow stops when O2and Ar deplete.
When all the gas in the burner is collected in trap 2,the residual gas in trap 1 will be released by heating the trap to about 200◦C.It contains all the krypton in the original sample,but being still mostly N2and O2, together with methane and some argon.The typical size is about 0.3 L(excluding methane).The gas released from trap 1 is also introduced to the burner with a fl ow rate controlled to be less than 50 mL/min.
When most N2,O2and CH4are removed in the burner,the residual gas is transferred to another activated liquid-N2cooled charcoal trap(trap 3,10 mL volume,1 g charcoal of 16−32 mesh).Typical time needed for the whole distillation and titanium reaction process is about 4 h for an air sample of 10 L.

FIG.3(a)Observed residual gas pressure in the“burner”during cryogenic distillation of an air sample of 10 L.(b)The measured fl ow rates of di ff erent gases.The total fl ow rate was controlled to be 50 mL/min by a mass fl ow controller. The distillation process was separated into two stages as indicated with vertical dotted lines.When the gas pressure in the“burner”is high,the gas fl ow is stopped for 20 min also and restarted when the argon gas is transferred to a cold trap.
C.Chromatographic separation of krypton
A gas chromatographic(GC)separation process is applied to extract krypton gas from the sample.The residual gas in trap 3 is released by heating the trap to 200◦C and fl ushed into a chromatographic column.The column is fi lled with a molecular sieve(MS 5A,grain size of No.60−80,diameter of 6 mm,length of 2 m) and installed in a constant temperature bath at 30◦C. Pure helium(99.999%purity,30 mL/min)is used as carrier gas.Characteristic elution peaks of various gas components are monitored with a thermal conductivity detector(TCD),and they are shown in Fig.4.Because several milliliters of argon and almost all krypton(micro-liters)are presented here,the chromatographic separation process includes a 2.5 min collection of argon and a 3 min collection of krypton,which is shown in Fig.4.

FIG.4 Chromatograms of the elution times of various gases originally extracted from an air-like sample.Two pairs of vertical dotted lines indicate the time ranges for Ar and Kr gas collection during 1st GC separation.The solid line shows the fi nal constituents of the Kr sample.
The collected argon gas,together with the gas previously stored in trap 2,is transferred into a chamber installed with a Ti-getter pump(Getter 1,500◦C,Nanjing Huadong Electronics Co.)to get rid of residual N2. After that,the argon gas is collected in a sample holder fi lled with activated charcoal at liquid-N2temperature, and then stored at room temperature.A second run of chromatographic separation is applied to extract the Kr gas,which is also shown in Fig.4.Then a getter process is applied to remove residual contaminants from the obtained krypton sample.Finally,the puri fi ed krypton gas is also collected in a sample holder fi lled with activated charcoal at liquid-N2temperature,being ready for ATTA measurement.The duration of the GC separation is about 1 h.
Note that the TCD signal in the GC process has been calibrated by using pure Ar,N2,Kr,and CH4samples. The areas under the chromatographic peaks are used to determine the contents of various components in the obtained krypton gas.The quantity of extracted argon is derived from the pressure gauge and the volume of the sample holder.Typically 90 mL argon can be obtained from an ambient air sample of 10 L(STP).
III.RESULTS AND DISCUSSION
A.Yield and purity of products
The e ffi ciency and purity of the extraction process were tested by several samples:ambient air samples with volumes varying from 1 L to 10 L(STP),two air samples of 10 L and mixed with 1%CH4,and two groundwater samples.The GC data of recovered krypton and argon gases are shown in Fig.5.The sizes of original samples and recovered Kr/Ar gases are presented in Table I.

FIG.5 Chromatograms of the fi nal components of(a)Kr and(b)Ar gases recovered from air samples of di ff erent sizes. The curve at the bottom of each panel is from a blank sample (pure helium).
Although Ar and N2contents are observed from the chromatography spectra(Fig.5(a)),which may result from air leak during the collection process.The size of “impurities”in obtained Kr samples becomes relatively larger when the original sample size gets smaller.Since only 1 ppm of ambient air is krypton,even at the worst case(1 L air sample,Fig.5(a)),the content of contaminated krypton relative to the whole krypton sample size is less than 0.1%and therefore negligible.
A small N2peak also presents in the TCD spectra of the recovered argon sample(Fig.5(b)).It indicates that the Ti-reaction and getter cannot completely remove N2from the argon sample.But the content of N2is below 1%.In addition,we cannot detect any loss of krypton(<0.1%)in the distillation process.Since the residual small impurities have no in fl uence on ATTA measurements,there is no need of further e ff orts to remove the impurities from the obtained krypton and argon samples.
For ambient air gases and CH4-rich samples,the yields of Kr and Ar are more than 85%and 90%,respectively.The O2-poor samples were extracted in fi eld from groundwater,and their initial constituents were analyzed by gas chromatography.Although original krypton contents in these two samples are too small

TABLE I The quality of processed gas(content of Kr inµL,content of Ar in mL),extraction yield(in%)and purity(in %)of Kr and Ara.
aThere is an uncertainty of 2.5%in the given yields of krypton and argon.
bCalculated values from krypton concentration of 1.14 ppm by volume in the earth’s atmosphere.cCalculated values from argon concentration of 0.934%.
dGas mixture with enriched CH4.
eDissolved gas samples extracted from groundwater. to be determined by conventional chromatography,the sizes of recovered krypton gases agree reasonably with the estimated values according to the solubility of krypton and the temperature of the groundwater.
B.Isotopic fractionation and environmental samples
The extracted krypton gas is ready for ATTA measurements.Figure 6 shows the fl uorescence signal of the trapped stable krypton isotopes when scanning the laser frequency.Two krypton samples were tested,one is a commercial pure krypton gas sample bought in 2007 (denoted as“2007 bottle”),and the other one is from a groundwater sample(the last sample shown in Table I). As shown in the fi gure,relative intensities of respective isotopes remain the same in both samples,indicating no detectable isotopic fractionation e ff ect in the gas extraction and puri fi cation process.When the laser frequency is set on resonance with the rare isotope85Kr or81Kr, image of single atoms will be detected by a sensitive EMCCD camera and counts of individual atoms will be used to derive the isotopic abundances[24].Two “O2-poor”samples extracted from deep groundwater obtained in north China plain are presented in Table I as application examples of the system.For the fi rst sample,5 counts of85Kr and 228 counts of81Kr have been obtained in 4 h.The isotopic abundances of85Kr and81Kr,relative to the modern values,are determined to be 0.3%and 71±5%,respectively,which leads to a81Kr age of about 113±23 ky.For the second sample, 18 counts of85Kr and 411 counts of81Kr have been recorded in 4 h.The relative-to-modern isotopic abundances of85Kr and81Kr are 0.6%and 106±6%,respectively.It indicates that the age of this groundwater sample is beyond both detection ranges of85Kr and81Kr,and should be older than 50 y.The very low85Kr counts also indicate that air contamination throughout the sample extraction and puri fi cation process is negligible.
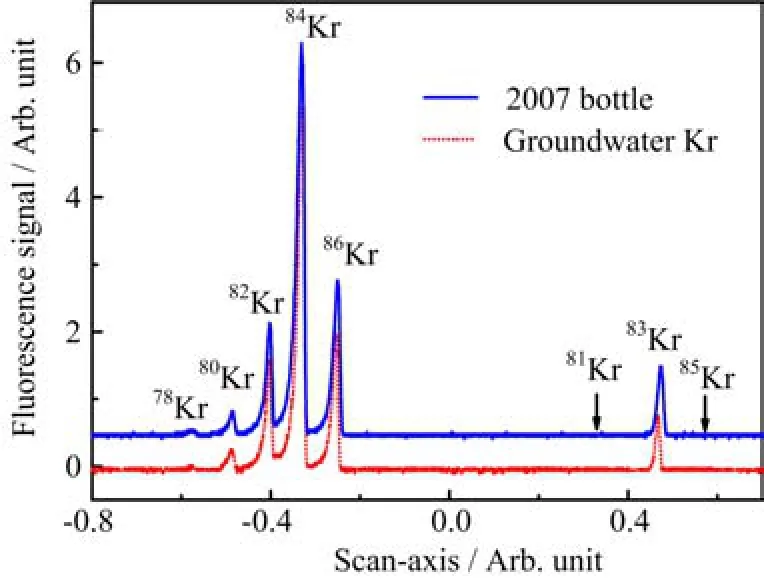
FIG.6 The fl uorescence spectra of krypton recovered from one groundwater sample and the“standard”krypton from a commercial gas bottle in 2007.The spectra show stable isotopes of krypton(78Kr,80Kr,82Kr,83Kr,84Kr and86Kr) and their relative abundances,and demonstrates that there is no signi fi cant isotopic fractionation throughout the whole krypton separation process.Two arrows mark the position of the two rare isotopes81Kr and85Kr.
IV.CONCLUSION
we have developed an apparatus to extract krypton and argon gases from air-like gas samples using a combination of cryogenic distillation,chemical absorption by titanium,and gas chromatography.A portable sampling instrument has also been developed to extract solved air from groundwater samples in the fi eld.The system has been tested by applying several di ff erent gas samples,including ambient air samples with a size of 1−10 L,synthesized CH4enriched samples which mimic gases extracted from groundwater,and also two samplesobtained from real groundwater in the fi eld.Krypton and argon gases can be separated with an e ffi ciency better than 90%.The system ful fi lls present needs of the ATTA measurement of the rare noble-gas isotopes including85Kr,81Kr and39Ar.
V.ACKNOWLEDGMENTS
This work was supported by the Special Fund for Land and Resources Research in the Public Interest (No.201511046)and the National Natural Science Foundation of China(No.21225314 and No.41102151).We would like to give our gratitude to Zong-yu Chen from IHEG for organizing the fi eld campaign.
[1]F.Vonhippel,D.H.Albright,and B.G.Levi,Sci.Am. 253,40(1985).
[2]K.Winger,J.Feichter,M.B.Kalinowski,H.Sartorius, and C.Schlosser,J.Environ.Radioact.80,183(2005).
[3]G.M.Yang,L.Y.Tu,C.F.Cheng,X.Y.Zhang,and S.M.Hu,Chin.J.Chem.Phys.28,445(2015).
[4]W.M.Jr.Smethie,D.K.Solomon,S.L.Schi ff,and G. G.Mathieu,J.Hydrol.130,279(1992).
[5]J.Held,S.Schuhbeck,and W.Rauert,Appl.Radiat. Isot.43,939(1992).
[6]B.Ekwurzel,P.Schlosser,W.M.Smethie Jr.,L.N. Plummer,E.Busenberg,R.L.Michel,R.Weppernig, and M.Stute,Water Resour.Res.30,1693(1994).
[7]R.Althaus,S.Klump,A.Onnis,R.Kipfer,R. Purtschert,F.Stauer,and W.Kinzelbach,J.Hydro. 370,64(2009).
[8]N.Momoshima,F.Inoue,S.Sugihara,J.Shimada,and M.Taniguchi,J.Environ.Radioact.101,615(2010).
[9]B.E.Lehmann,H.H.Loosli,D.Rauber,N.Thonnard, and R.D.Willis,Appl.Geochem.6,419(1991).
[10]P.Collon,W.Kutschera,H.H.Loosli,B.E.Lehmann, R.Purtschert,A.Love,L.Sampson,D.Anthony,D. Cole,B.Davids,D.J.Morrissey,B.M.Sherrill,M. Steiner,R.C.Pardo,and M.Paul,Earth Planet.Sci. Lett.182,103(2000).
[11]B.E.Lehmann,A.Love,R.Purtschert,P.Collon,H. H.Loosli,W.Kutschera,U.Beyerle,W.Aeschbach-Hertig,R.Kipfer,S.K.Frape,A.Herczeg,J.Moran,I. N.Tolstikhin,and M.Groning,Earth Planet.Sci.Lett. 211,237(2003).
[12]N.C.Sturchio,X.Du,R.Purtschert,B.E.Lehmann, M.Sultan,L.J.Patterson,Z.T.Lu,P.M¨uller, T.Bigler,K.Bailey,T.P.O’Connor,L.Young,R. Lorenzo,R.Becker,Z.El Alfy,B.El Kaliouby,Y.Dawood,and A.M.A.Abdallah,Geophys.Res.Lett.31, L05503(2004).
[13]C.Buizert,D.Baggenstos,W.Jiang,R.Purtschert,V. V.Petrenko,Z.T.Lu,P.M¨uller,T.Kuhl,J.Lee,J.P. Severinghaus,and E.J.Brook,Proc.Natl.Acad.Sci. USA 111,6876(2014).
[14]H.H.Loosli,Earth Planet.Sci.Lett.63,51(1983).
[15]H.H.Loosli,M.M¨oll,H.Oeschger,and U.Schotterer, Nucl.Instrum.Meth.B 17,402(1986).
[16]P.Collon,M.Bichler,J.Caggiano,L.D.Cecil,Y.El Masri,R.Golser,C.L.Jiang,A.Heinz,D.Henderson, W.Kutschera,B.E.Lehmann,P.Leleux,H.H.Loosli, R.C.Pardo,M.Paul,K.E.Rehm,P.Schlosser,R. H.Scott,W.M.Smethie Jr.,and R.Vondrasek,Nucl. Instrum.Meth.B 223,428(2004).
[17]J.A.Corcho Alvarado,R.Purtschert,F.Barbecot, C.Chabault,J.Rueedi,V.Schneider,W.Aeschbach-Hertig,R.Kipfer,and H.H.Loosli,Water Resour.Res. 43,W03427(2007).
[18]N.Aoki and Y.Makide,Chem.Lett.34,1396(2005). [19]J.Ahlswede,S.Hebel,J.O.Ross,R.Schoetter,and M. B.Kalinowski,J.Environ.Radioact.115,34(2013).
[20]H.H.Loosli and H.Oeschger,Earth Planet.Sci.Lett. 7,67(1969).
[21]P.Collon,T.Antaya,B.Davids,M.Fauerbach,R. Harkewicz,M.Hellstrom,W.Kutschera,D.Morrissey, R.Pardo,M.Paul,B.Sherrill,and M.Steiner,Nucl. Instrum.Meth.B 123,122(1997).
[22]C.Y.Chen,Y.M.Li,K.Bailey,T.P.O’Connor,L. Young,and Z.T.Lu,Science 286,1139(1999).
[23]W.Jiang,K.Bailey,Z.T.Lu,P.Mueller,T.P. O’Connor,C.F.Cheng,S.M.Hu,R.Purtschert,N. C.Sturchio,Y.R.Sun,W.D.Williams,and G.M. Yang,Geochim.Cosmochim.Acta 91,1(2012).
[24]G.M.Yang,C.F.Cheng,W.Jiang,Z.T.Lu,R. Purtschert,Y.R.Sun,L.Y.Tu,and S.M.Hu,Sci. Rep.3,1(2013).
[25]L.Y.Tu,G.M.Yang,C.F.Cheng,G.L.Liu,X.Y. Zhang,and S.M.Hu,Anal.Chem.86,4002(2014).
[26]W.Jiang,W.Williams,K.Bailey,A.M.Davis,S.M. Hu,Z.T.Lu,T.P.O’Connor,R.Purtschert,N.C. Sturchio,Y.R.Sun,and P.Mueller,Phys.Rev.Lett. 106,103001(2011).
[27]F.Ritterbusch,S.Ebser,J.Welte,T.Reichel,A.Kersting,R.Purtschert,W.AeschbachHertig,and M.K. Oberthaler,Geophys.Res.Lett.41,6758(2014).
[28]Z.T.Lu,P.Schlosser,W.M.Smethie Jr.,N.C.Sturchio,T.P.Fischer,B.M.Kennedy,R.Purtschert,J. P.Severinghaus,D.K.Solomon,T.Tanhua,and R. Yokochi,Earth-Sci.Rev.138,196(2014).
[29]R.F.Weiss,Deep-Sea Res.17,721(1970).
[30]R.F.Weiss and T.K.Kyser,J.Chem.Eng.Data 23, 69(1978).
[31]W.M.Smethie Jr.and G.Mathieu,Mar.Chem.18,17 (1986).
[32]T.Mohamed,J.Strohaber,R.Nava,A.Kolomenskii, N.Thonnard,and H.A.Schuessler,J.Am.Soc.Mass Spectrom.23,1260(2012).
[33]R.Yokochi,L.J.Heraty,and N.C.Sturchio,Anal. Chem.80,8688(2008).
[34]A.Gabelman and S.T.Hwang,J.Membr.Sci.159,61 (1999).
[35]P.C.Probst,R.Yokochi,and N.C.Sturchio,4th Mini Conference on Noble Gases in the Hydrosphere and in Natural Gas Reservoirs,(2007).
[36]B.Loose,M.Stute,P.Alexander,and W.M.Smethie, Water Resour.Res.45,W00D34(2009).
[37]T.Ohta,Y.Mahara,N.Momoshima,F.Inoue,J.Shimada,R.Ikawa,and M.Taniguchi,J.Hydro.376,152 (2009).
[38]T.Matsumoto,L.F.Han,M.Jaklitsch,and P.K.Aggarwal,Groundwater 51,461(2013).
Chinese Abstracts(中文摘要)
时间分辨偏振红外光谱及其应用.................................1张文凯∗(北京师范大学高等量子研究中心,物理系,北京100875)
摘要:时间分辨偏振红外光谱已被广泛应用于研究光化学过程中的分子结构动力学.通过测定瞬态物质跃迁偶极矩之间的角度等结构信息,可以提供光化学过程中伴随的电荷分布、分子结构和构象变化等动态信息.包括简要介绍时间分辨偏振红外光谱技术的原理和应用:(i)时间分辨偏振红外光谱概述;(ii)时间分辨偏振红外光谱的原理及其优势;(iii)利用时间分辨偏振红外光谱探测多种化学动力学过程,例如蛋白质构象动力学、激发态的电子局域化和光致异构化等;(iv)时间分辨偏振红外光谱的局限和发展前景. 利用基质隔离红外光谱结合理论计算,研究了激光溅射获得的第五族金属原子和硫化氢分子的反应.结果表明金属原子插入H2S的H−S化学链形成HMSH分子(M=V,Nb,Ta). 对Nb和Ta该HMSH分子重排为H2MS分子.HMSH分子和H2S进一步反应生成H2M(SH)2分子.通过S同位素标定确定了产物的分子结构,同时我们用DFT(B3LYP和BPW91)理论计算预测了产物分子的能量、结构和振动频率.通过DFT IRC计算研究了第五族金属原子和2S分子的反应机理.HVSH分子通过光照解离为VS和H2,然后通过退火可以发生VS和H2复合反应.计算表明HVSH释放H2需要16.9 kcal/mol的活化能及吸热13.5 kcal/mol. 采用共振拉曼光谱学和完全活化空间自洽场方法研究了苯基叠氮被激发到S2(A')、S3(A')和S6(A')光吸收态后的结构动力学.基于傅立叶变换拉曼、傅立叶变换红外、紫外、密度泛函计算和简正模式分析,指认了紫外吸收光谱和振动光谱.获得了环己烷、乙腈和甲醇溶剂中273.9、252.7、245.9、228.7、223.1和208.8 nm等不同激发波长下的A、B和C带共振拉曼光谱,以探测Franck-Condon区域的结构动力学.CASSCF计算获得了单重电子激发态能量最低点和势能面交叉点的电子激发能和优化几何结构.结果表明,苯基叠氮在S2(A')、S3(A')和S6(A')态上的激发态结构动力学各不相同.与Kasha规则相符,S2S1(1)和S2S1(2)势能面交叉点在S2(A')激发态衰变动力学和N7=N8链解离中扮演着重要角色.提出了两条主要衰减通道:S2,min→S0辐射通道和S2,FC(ππ∗)→S2(ππ∗)/S1(nπ∗)→S1(nπ∗)非辐射通道. 利用团簇模型研究了质子化水团簇对乙炔的溶剂化作用. H+(C2H2)(H2O)n(n=1~5)的量子化学计算结果表明,水分子倾向与乙炔的π电子形成新型OH···π氢链作用,并且乙炔的第一溶剂层需要4个水分子来完成.模拟的红外光谱揭示了OH···π氢链作用后的OH伸缩振动是研究乙炔与水溶剂化过程的灵敏探针.这些红外光谱可以用红外光解离光谱实验方法测得,将为理解OH···π氢链作用以及质子化水团簇如何溶剂化乙炔提供有力的科学数据. 利用高精度的CASSCF和MS-CASPT2电子结构计算方法系统地研究了2-(2'-羟基苯基)-4-甲基噁唑的光物理和光化学机理. 在CASSCF级别,首先优化得到势能面极小结构和圆锥交叉结构,及激发态质子转移、异构化、和失活的极小能量路径.然后用MS- 利用脉冲激光溅射-超声分子束载带方法制备气相硼羰基络合物正离子.采用红外光解离光谱研究了、和B2(CO)4+的振动光谱.研究结果表明具有非常强的B−CO链,无法直接获得其红外光解离光谱.对B(CO)4+的光解离光谱研究表明该离子是一个B(CO)3+和CO之间弱相互作用络合物.其中B核具有平面D3h对称性结构,中心硼具有稳定的8电子组态.具有平面的D2h对称性结构,其中的B−B链包含一个σ链和半个π链.自然轨道能量分解分析(EDA-NOCV)表明在B(CO中的B−CO成链作用中OC→B(σ)要比B→CO(π)反馈作用强. 利用飞秒时间分辨质谱技术研究了邻碘甲苯分子在266 nm激发下的光解动力学.光解产物碎片通过800 nm强激光场下的多光子电离实现探测.拟合光解产物C7H7自由基和I原子随泵浦-探测延迟时间变化的信号,得到解离时间为380±50 fs,它反映的是266 nm同时激发nσ∗和ππ∗态后C−I链的平均解离时间.此外,还利用基态碘原子的共振波长298.23 nm作为探测光,通过共振增强多光子电离方法对解离生成的基态碘原子进行了选择性探测.拟合I+随泵浦-探测延迟时间变化的信号,得到解离时间为400±50 fs,这与通过800 nm多光子电离得到的解离时间一致,表明解离生成的主要产物是基态碘原子. 通过采用真空紫外(VUV)激光速度-地图成像-TPE(真空紫外VMI-TPE)方法获得了高分辨率初始光电子(TPE)氯苯)的光谱,炔丙基自由基)和烯丙基.观察到的真空紫外VMI-TPE方法的光电子能量分辨率在1~2 cm−1,可以和在真空紫外激光脉冲场电离光电子(VUV-PFI-PE)的测量媲美.类似真空紫外PFI-PE测量,真空紫外VMI-光电子(真空紫外VMI-PE)和真空紫外VMI-TPE测量能量分辨率依赖于直流电场在光电离区加速电子.C6H5Cl和C3H3的电离初始值的降低为F的函数表示Stark偏移校正为VUV-VMI-TPE测量由−3.1√ 报道和频振动光谱在交叉传播的实验构型下的理论公式推导和实验结果.在交叉传播的和频振动光谱实验室中,可见光和红外光通过相互垂直的入射面同时照射在界面上,从而避免了对使用同时能够透过可见和红外激光束的光学元件的要求.这种交叉实验构型能够直接应用到封闭在真空或者压力腔体中的界面,使得在用远红外直接探测金属氧化物及其它低频界面振动模式实验的窗口材料有更多的选择. 利用超快泵浦探测红外光谱、稳态线性红外光谱和计算化学方法,对过渡金属羰基化合物Mn(CO)5Br和Re(CO)5Br的振动和结构动力学进行了研究.借助羰基的两个伸缩振动峰(处于低频的A1模式和处于高频的简并E模式)进行了观测.结果表明,在两个配合物中,A1和E模式振动峰的振动频率位置及频率差都与中心金属原子对羰基的链级和振动力常数的影响相关.而A1模式比E模式的线宽宽一些,部分由于振动寿命的影响.此外,从瞬态光谱中获得了振动模式依赖的对角非谐性常数,发现在两个羰基化合物中E模式的非谐性总是较小. 电化学反应在许多学科中扮演着很重要的角色.对于化学分析来说,振动光谱是一门很好的原位测量技术,然而由于红外光在电极和电解质中强烈的衰减使得它在电化学反应中的应用受到了很大的限制.本文中论证了在金属电极上结合适当的亚波长等离子体结构,在OH伸缩振动频段内,通过激发表面等离激元能够大大增强金属与液态水电化学界面的红外场强.这对于原位振动光谱的研究,特别是当用到表面灵敏的光学混频技术时可产生很大的促进作用. 采用时间分辨红外吸收光谱方法,研究了邻甲基苯甲酸安息香酯在266 nm激光作用下发生的光化学反应,并考察了溶剂对反应过程的影响.从瞬态红外光谱上观察到了邻甲基苯甲酸和苯甲酰自由基及苯偶酰,提供了直接的光谱证据证明了脱保护反应及C−C=O处均裂反应的发生.通过对比溶剂中不同水含量下均裂反应中间体及邻甲基苯甲酸的产率,发现水含量在溶剂中的增加能促进脱保护反应的发生.关链词:安息香,笼化合物,光致脱保护反应,时间分辨红外吸收光谱 利用新建激光溅射交叉分子束装置,结合时间切片速度成像技术开展了金属原子态-态反应动力学的相关研究.超声金属原子束是由激光溅射金属棒产生,结合无气体溢流通道的自由扩散设计,得到了质量很好的金属原子超声束.本文选择Al+O2反应体系来测试新建金属交叉分子束实验装置的性能.通过(1+1)共振多光子电离技术,以AlO(D2Σ+)为中间态来探测特定转动态的产物AlO自由基.相同波长下可以同时得到反应产物AlO(X2Σ+,ν=0,N和N+14)两个转动态的速度成像,分别对应着∆ν=1的P(N)和R(N+14)跃迁. 在244.145 nm同时探测到P(15)和R(29)的跃迁,形成的两个环在切片成像图中可以完全区分开,这两个跃迁分别对应着反应产物AlO(ν=0,N=15)和AlO(ν=0,N=29)两个转动态.对应此两个转动态的能级差为403 cm−1.这两个反应产物转动态的区分表明了该实验装置与最近的一篇研究报道[J.Chem.Phys.140,214304 (2014)]相比较,具有较好的碰撞能量分辨率. 通过双光子光电子的方法探测了TiO2(011)-(2×1) 和TiO2(110)-(1×1)表面的光催化氧化甲醇的性质.在吸附了甲醇的二氧化钛(011)和(110)界面处探测到了一个费米能级以上2.5 eV的电子激发态,该电子激发态可作为测试二氧化钛界面还原性的探针使用.利用此探针在甲醇/TiO2(011)-(2×1)和甲醇/TiO2(110)-(1×1)界面探测到了一个随光照时间的电子激发态信号变化,这一变化可以归于光催化生成的表面羟基对界面还原性的影响.由此得出的光催化氧化甲醇的速率TiO2(110)-(1×1)比TiO2(011)-(2×1)快了大约11.4倍.这可能由于表面原子结构排布的原因不同.本工作不仅介绍了一个利用双光子光电子能谱探测到的甲醇/TiO2界面电子结构的细节特征,还揭示了表面结构对二氧化钛光反应性质的重要影响. 在量子动力学计算中,有时候为了规避奇点问题或者节省计算量,我们经常需要对哈密顿量进行变换.然而,在使用傅里叶基矢计算时,哈密顿量的变换形式容易导致哈密顿矩阵失去厄米性,进而有些情况下使数值计算变得不稳定.本文主要讨论构建具有厄米性的哈密顿算符的方法.以三原子分子为例,构建了链长—链角和Radau坐标下描述分子运动的各种形式的哈密顿量.基于这些哈密顿量,采用含时波包方法计算了OClO分子的吸收光谱,讨论了非厄米性矩阵对计算结果的影响.本文所得到的结论对基于基函数展开的量子动力学计算都是适用的. 利用光电子能谱及密度泛函理论计算对TiGen−(n=7~12)团簇的几何结构及电子特性等进行了系统研究.对于TiGen−负离子及中性TiGen,在n=8时出现了钛原子半内嵌的船型结构;在n=9~11时,新增的锗原子加盖到这种船型结构上,逐步形成钛原子完全内嵌的结构.TiGe12−团簇具有一种钛原子内嵌的变形六棱柱结构.自然布居分析结果显示,对于n=8~12的TiGen−/0团簇,随着内嵌结构的形成,有电子从锗原子转移到钛原子,说明其电荷转移方式与结构演变密切相关. 蛋白质的酰胺A谱带对蛋白质的酰胺氢链结构很敏感.然而由于该谱带和水的OH伸缩振动谱带严重重叠,导致在蛋白质水溶液中原位测量酰胺A谱带依旧很困难.我们提出了一种新的分析方法用于原位测量水溶液中的酰胺A谱带.这个方法称为拉曼除谱法.将蛋白质水溶液光谱除以纯水光谱即可获得拉曼除谱.利用数值模拟从数学上肯定了使用拉曼除谱可以直接获得酰胺A谱带.我们还通过测量溶菌酶和α-糜蛋白酶的固体和水溶液的拉曼光谱,这些光谱也证实了可以通过拉曼除谱法直接获取酰胺A谱带.利用拉曼除谱还分析了溶菌酶的热变性过程.这些研究表明拉曼除谱可以原位地表征水溶液中的蛋白质酰胺A谱带. 利用激光闪光光解技术研究了蒽醌-2-磺酸钠(AQS)在吡啶离子液体N-丁基吡啶四氟硼酸盐([BPy][BF4])与水(H2O)混合体系中的光化学反应过程.实验结果表明,AQS的激发三重态(3AQS∗)会与H2O快速反应,不断增加[BPy][BF4]在混合体系中的体积比(VIL),瞬态吸收光谱发生了很大变化.510 nm附近的瞬态吸收带变化最大,在0<VIL<0.1时,吸光度会随着[BPy][BF4]的增加而增加;而在VIL>0.1时,吸光度则随着比例的增加而减小.然而380 nm附近吸收带的吸光度却一直在增加.通过拟合近似地得到了瞬态物种B和3AQS∗的表观动力学参数.另外还讨论了3AQS∗与阳离子之间的夺氢反应,通过对350~420 nm处光谱图的分析,推断出这一范围的瞬态吸收光谱是3AQS∗与AQSH·的叠加谱.在混合体系 利用荧光非共线光参量放大光谱技术测量了DCM染料乙醇溶液的溶剂化动力学过程.实验结果表明,瞬态荧光光谱经过光谱矫正后,可以产生准确的溶剂化相关函数以及溶剂化过程中瞬态光谱峰值频率移动.本文的工作表明荧光非共线光参量放大光谱技术有益同时关注荧光强度动力学以及光谱谱型演化的研究领域. 放射性惰性气体同位素85Kr(半衰期为10.8年)、39Ar(半衰期为269年)和81Kr(半衰期为22.9万年)是理想的环境示踪剂,基于激光技术的原子阱痕量分析方法(Atom trap trace analysis,ATTA)可以实现对空气、地下水等环境样品中这几种同位素的有效探测.在进行ATTA测量之前,需要将样品中的氪气和氩气有效分离出来.利用低温蒸馏、海绵钛化学吸附和气相色谱分离等技术,可以从1~10 L气体样品中分别提取出90%以上的氪气和98%以上的氩气,从而满足ATTA测量的样品要求.通过对包括两个野外地下水样品等一系列样品进行分离实验,验证了气体分离装置的可靠性能.
关链词:超快光谱,红外光谱,偏振,时间分辨红外光谱
基质隔离红外光谱结合理论计算研究第五族金属原子和硫化氢反应. ..............................................................11赵杰,许兵,俞文杰,王雪峰∗(同济大学化学系,上海市化学品分析、风险评估及控制重点实验室,上海200092)
关链词:硫化氢,基质隔离,过渡金属,DFT理论计算
苯基叠氮在光吸收激发态上的结构动力学−共共振拉曼和量子力学计算研究.......................................................21袁荣单,薛佳舟,郑旭明∗(浙江理工大学化学系,杭州310018)
关链词:苯基叠氮,结构动力学,衰减动力学,共振拉曼光谱,CASSCF计算,锥形交叉点
质子化水团簇对乙炔溶剂化作用的结构与红外光谱.............31孔祥涛a,雷鑫a,袁勤勤a,张冰冰a,b,赵志a,c,杨冬a,蒋述康a,戴东旭a,江凌a∗(a.中国科学院大连化学物理研究所,分子反应动力学国家重点实验室,能源材料化学协同创新中心,大连116023;b.大连理工大学,精细化工国家重点实验室,大连116024;c.大连理工大学,三束材料改性教育部重点实验室,大连116024)
关链词:乙炔,水,溶剂化,红外光解离光谱,量子化学计算
噁唑体系激发态质子转移和失活的理论研究...................38谢斌斌,李春香,崔刚龙∗,方遒∗(北京师范大学化学学院,理论与计算光化学教育部重点实验室,北京100875)
CASPT2方法对所有得到的结构和能量路径进行单点能量校正,我们发现在含有OH···N氢链的构象异构体中,激发态质子转移基本上是一个无垒的过程;在含OH···O氢链的构象异构体中,激发态质子转移被抑制了.此外,找到两个能量较低的酮式S1/S0圆锥交叉结构,使得激发态质子转移生成的S1酮式结构可以很快失活到达基态.但是,醇式S1/S0圆锥交叉结构能量较高,抑制了S1醇式结构的激发态失活.
关链词:激发态质子转移,光异构化,圆锥交叉,从头算,光化学
关链词:硼碳基络合物,σ-π配链,红外光解离光谱,理论计算
邻碘甲苯分子光解动力学的飞秒时间分辨质谱研究..............53刘志明,王艳梅,胡春龙,龙金友,张冰∗(中国科学院武汉物理与数学研究所,波谱与原子分子物理国家重点实验室,武汉430071)
关链词:邻碘甲苯,光解,解离时间,飞秒时间分辨质谱
高分辨率初始光电子能谱由真空紫外激光速度—映射图像的方法... ..............................................................59律洲,高蕻,徐运涛,杨磊,林周成,Yanice Benitez,伍灼耀∗(加州大学戴维斯分校化学系,戴维斯95616)
F管辖,这是半经典预测值−6.1√ F的一半.我们还测量C6H5Cl和C3H5的真空紫外光能量的真空紫外VMI-PE谱接近其电离初始值.在VUV-VMI-PE测量中观察到的阳离子振动谱和振动级数,.真空紫外VMI-TPE可以实现更高的实验灵敏度和类似真空紫外PFI-PE测量的能量分辨率,使真空紫外VMITPE法成为高分辨率真空紫外PFI-PE测量一个很好的替代.
关链词:光电离,初始光电子,速度-地图成像,自由基
交叉传播构型的和频振动光谱.................................70付力a,陈顺利a,b,干为b,王鸿飞a∗(a.美国能源部西北太平洋国家实验室环境分子科学研究所,里奇兰99352;b.中国科学院新疆理化技术研究所环境科学和技术实验室,乌鲁木齐830011)
Chin.J.Chem.Phys.Vol.29,No.1
化学物理学报,第29卷,第1期
这一交叉实验构型的潜在应用包括表面科学、材料科学、基础催化科学以及低温下的分子科学等方面.
关链词:和频产生,振动光谱,共向传播,反向传播,交叉传播
过渡金属羰基化合物Mn(CO)5Br和Re(CO)5Br结构与动力学的超快光谱.....................................................81封敏军a,b,杨帆a,王建平a∗(a.中国科学院化学研究所北京分子科学国家实验室,分子反应动力学实验室,北京100190;b.中国科学院大学,北京100149)
关链词:过渡金属羰基化合物,瞬态红外光谱,振动弛豫,非谐性
金属/水界面振动光谱的等离基元场增强.......................87刘志华,徐倩,刘韡韬∗(复旦大学物理系,应用表面物理国家重点实验室,微纳光子结构教育部重点实验室,先进微结构协同创新中心,上海200433)
关链词:金属-水界面,表面等离子体激发,光学异常透射
安息香笼化合物光化学反应的时间分辨红外光谱................91代小娟a,余友清a,刘坤辉b∗,苏红梅a,b∗(a.中国科学院化学研究所,北京分子科学国家实验室(筹),北京100190;b.北京师范大学化学学院,北京100875)
利用激光溅射原子束装置结合时间切片速度成像技术对于金属态-态反应动力学的相关研究........................................99董常武,刘嘉兴,李芳芳,王凤燕∗(复旦大学化学系,化学能源材料协同创新中心,上海200433)
关链词:时间切片速度成像,交叉分子束,激光溅射,金属原子反应动力学
甲醇在二氧化钛上的光化学性质与表面结构的相关性..........105郝群庆a,王志强a,毛新春b,周传耀a∗,戴东旭a,杨学明a∗(a.中国科学院大连化学物理研究所分子反应动力学国家重点实验室,大连
关链词:二氧化钛,电子激发态,双光子光电子能谱,光催化氧化速率
链长-链角和Radau坐标下哈密顿算符在傅里叶基组表象下的厄米性...........................................................112于德权a,黄鹤a,b,Gunnar Nymanc,孙志刚a,d∗(a.中国科学院大连化学物理研究所分子反应动力学国家重点实验室,大连116023;b.辽宁师范大学物理与电子技术学院,大连116029;c.瑞典哥德堡大学化学学院,哥德堡;d.中国科学与技术大学,量子信息与量子科技前沿协同创新中心,合肥230026)
关链词:哈密顿量,快速傅里叶变换,傅里叶基组,含时波包方法,吸收光谱
TiGen−(n=7~12)团团簇的光电子能谱及密度泛函理论研究...123邓晓娇,孔祥玉,徐西玲,许洪光∗,郑卫军∗(中国科学院化学研究所,北京分子科学国家实验室(筹),分子反应动力学国家重点实验室,北京100190)
关链词:光电子能谱,密度泛函理论,锗团簇
拉曼除谱原位测量水溶液中蛋白质的酰胺A谱带...............129汤城骞a,林珂b∗,周晓国a,c,刘世林a∗(a.中国科学技术大学合肥微尺度物质科学国家实验室,化学物理系,合肥230026;b.西安电子科技大学物理与光电工程学院,西安710071;c.量子信息与量子科技前沿协同创新中心,合肥230026)
关链词:拉曼除谱,酰胺A谱带,原位,蛋白质,水
高分辨的一氧化二氮光解离实验研究..........................135俞盛锐a,b,袁道福a,陈文韬a,谢婷a,王思雯a,杨学明a,b∗,王兴安a,c∗(a.中国科学技术大学化学与材料科学学院化学物理系物理化学高等研究中心,合肥230026;b.中国科学院大连化学物理研究所分子反应动力学国家重点实验室,大连116023;c.能源材料化学协同创新中心,合肥230026)
Chin.J.Chem.Phys.Vol.29,No.1
化学物理学报,第29卷,第1期
子在134.20、135.20和136.43nm波长下的真空紫外光解动力学.实验中通过采集解离产物O(1SJ=0)的离子影像来研究O(1SJ=0)+N2(X1Σ+g)这一解离通道.从各个波长下的实验影像可获得产物N2(X1Σ+g)的振动态分辨的结构,进而得到产物的总平动能谱和产物N2的振动态布居.实验结果表明在实验的光解波长下,产物N2(X1Σ+g)主要布居在v=2和v=3.此外,还得到了产物N2的振动态分辨的各向异性参数β,从中发现产物N2的β值在三个解离波长下均表现出相似的特征,即随着振动量子数的增大,β值从趋近于2逐渐减小至1.4.这一现象表明低振动态产物是通过一个以平行跃迁解离为主的解离过程产生的,而高振动态的产物来自于一个更加弯曲的中间构型的解离.此推论与在平动能谱中所见到的最强转动态布居随着振动量子数的增大而出现的位移是相一致的.
关链词:一氧化二氮,离子成像,真空紫外,光解
吡啶离子液体与水混合体系中蒽醌-2-磺酸钠的激光闪光光解机理.. .............................................................140朱光来a∗,张良伟a,刘艳成b,崔执凤a,许新胜a,吴国忠b∗(a.安徽师范大学原子与分子物理研究所,芜湖241000;b.中国科学院上海应用物理研究所,上海201800)
中,3AQS∗分别与H2O和[BPy][BF4]的反应是一对竞争反应.还发现在高浓度的离子液体环境下,体系的整体反应速率会减弱.
关链词:激光闪光光解,蒽醌-2-磺酸钠,离子液体,瞬态吸收,夺氢基于荧光非共线光参量放大光谱技术的溶剂化动力学研究中的光谱矫正........................................................147党伟a,b,白晶晶a,张连水a∗,翁羽翔b∗(a.河北省光电信息材料重点实验室,河北大学物理科学与技术学院,保定071002;b.中国科学院物理研究所软物质物理重点实验室,北京100190)
关链词:原子阱痕量分析,气相色谱分离,放射性惰性气体
Supporting Information
Spectrum correction in the study of solvation dynamics by
fluorescence non-collinear optical parametric amplification
spectroscopy
Wei Danga,b, Jing-Jing Baia, Lian-Shui Zhanga∗, Yu-Xiang Wengb*
a Hebei Key Lab of Optic-electronic Information and Materials, College of Physics Science and Technology,
Hebei University, Wusi East 180, Baoding, 071002, China
bKey Laboratory of Soft Matter Physics, Institute of Physics, Chinese Academy of Sciences, No.8, 3rd
South Street, Zhongguancun, Beijing, 100190, China
E-mail address: zhangls@hbu.edu.cn; yuxiangweng@iphy.ac.cn
The detailed description of FNOPAS setup
The setup of FNOPAS is schematically presented In Figure S1. A laser pulse(150 fs, 300 uJ , at 800 nm and 1 kHz repetition rate) is split into two beams with a 1:1 energy ratio. The reflected fundamental beam passes through a delay line, then an inverted telescope consisting of a lens pair L2/L3 with a focal length of +200 mm and - 75 mm, respectively. This fundamental beam is frequency doubled to 400 nm in a BBO crystal (cutting angle 29.2°) of 1 mm thickness. This second-harmonic(SH) generation beam works as both the pump source for parametric amplification process and the gating beam of fluorescence photons. The transmitted fundamental beam is also frequency doubled to 400 nm in another BBO crystal (1mm, cutting angle 29.2°). After being focused by a lens L1(f=75 mm), this SH excites DCM ethanol solution (3×10-4mol/L). To suppress the population of triplet state, the solution is continuously stirred by a tiny magnetic bar during the measurement. The fluorescence from DCM solution is collected and imaged onto a 2 mm thick BBO crystal (cutting angle 31.5°) by a lens L4(f= 38.1 mm). A 500 nm long pass filter (F1) is placed to remove the scattered light of excitation beam. The fluorescence is gated and amplified through non-collinear optical parametric amplification process in the BBO crystal. The amplified fluorescence is recorded as the time-resolved spectrum by a CCD spectrograph. The details of data acquiring procedure have been given in the reference 1. Before data analysis, the acquired transient fluorescence spectra should be corrected with the curve of spectral gain. Due to transmission through ethanol solution, sample cell windows, lenses and the filter, fluorescence photons are subjected to group velocity dispersion (GVD). Thus GVD correction for the transient fluorescence spectra should be carried out after the spectral gain correction.
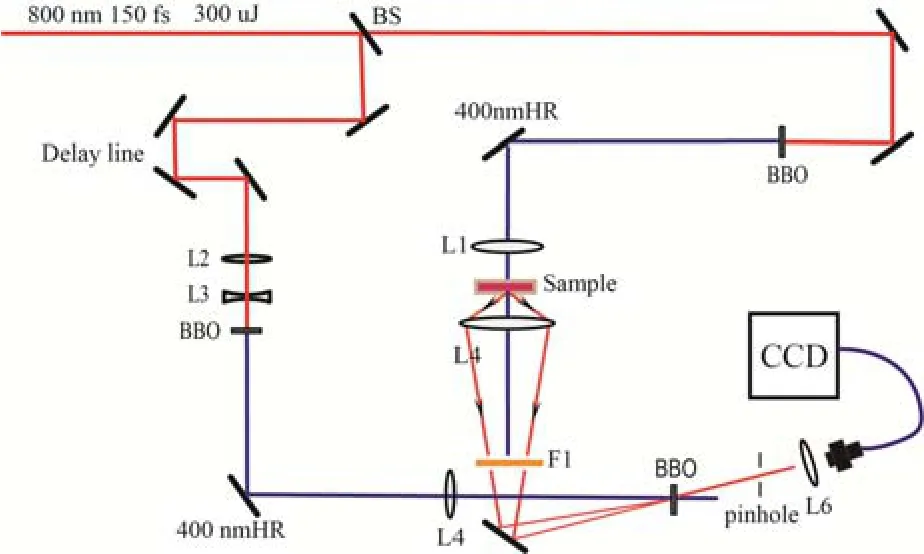
Fig.S1 The setup of femtosecond time-resolved fluorescence non-collinear optical parametric amplification spectroscopy. BS: 1:1 beam splitter; L1-L6: lens; F1: 500 nm long pass filter; 400nm HR: high reflectivity mirror at 400 nm; CCD: CCD spectrograph.
关链词:瞬态荧光光谱,溶剂化动力学,非共线光参量放大,光谱矫正
从环境样品中分离氩气和氪气用于放射性惰性气体探测........151涂乐义a,杨国民a,张向阳a,b,胡水明a∗(a.中国科学技术大学合肥微尺度物质国家实验室(筹),合肥230026;b.中国地质科学院水文地质环境地质研究所,石家庄050061)
Corresponding author.
猜你喜欢
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- ARTICLE Spectrum Correction in Study of Solvation Dynamics by Fluorescence Non-collinear Optical Parametric Ampli fi cation Spectroscopy†
- REVIEW Polarization Dependent Time-Resolved Infrared Spectroscopy and Its Applications†
- ARTICLE Reactions of Group V Metal Atoms with Hydrogen Sul fi de:Argon Matrix Infrared Spectra and Theoretical Calculations†
- ARTICLE Structural Dynamics of Phenyl Azide in Light-Absorbing Excited States: Resonance Raman and Quantum Mechanical Calculation Study†
- ARTICLE Structural and Infrared Spectroscopic Study on Solvation of Acetylene by Protonated Water Molecules†
- ARTICLE Excited-State Proton Transfer and Decay in Hydrogen-Bonded Oxazole System:MS-CASPT2//CASSCF Study†