ARTICLE Photochemical Reaction of Benzoin Caged Compound:Time-Resolved Fourier Transform Infrared Spectroscopy Study†
2016-04-08XiojunDiYouqingYuKunhuiLiuHongmeiSuBeijingNtionlLortoryforMoleculrSciencesInstituteofChemistryChineseAcdemyofSciencesBeijing100190ChinCollegeofChemistryBeijingNormlUniversityBeijing100875ChinDtedReceivedonDecemer23
Xio-jun Di,You-qing Yu,Kun-hui Liu∗,Hong-mei Su,∗.Beijing Ntionl Lortory for Moleculr Sciences,Institute of Chemistry,Chinese Acdemy of Sciences,Beijing 100190,Chin.College of Chemistry,Beijing Norml University,Beijing 100875,Chin(Dted:Received on Decemer 23,2015;Accepted on Jnury 25,2016)
ARTICLE Photochemical Reaction of Benzoin Caged Compound:Time-Resolved Fourier Transform Infrared Spectroscopy Study†
Xiao-juan Daia,You-qing Yua,Kun-hui Liub∗,Hong-mei Sua,b∗
a.Beijing National Laboratory for Molecular Sciences,Institute of Chemistry,Chinese Academy of Sciences,Beijing 100190,China
b.College of Chemistry,Beijing Normal University,Beijing 100875,China
(Dated:Received on December 23,2015;Accepted on January 25,2016)
The benzoin group caged compound has received strong interests due to its excellent photodeprotection properties and wide use in chemical and biological studies.We used timeresolved infrared spectroscopy to investigate the photochemical reaction of the benzoin caged compound,o-(2-methylbenzoyl)-DL-benzoin under 266 nm laser irradiation.Taking advantage of the speci fi c vibrational marker bands and the IR discerning capability,we have detected and identi fi ed the uncaging product 2-methylbenzoic acid,and two intermediate radicals of benzoyl and 2-methylbenzoate benzyl in the transient infrared spectra.Our results provide spectral evidence to support the homolytic cleavage reaction of C−C=O bond in competition with the deprotection reaction.Moreover,the product yields of 2-methylbenzoic acid and benzoyl radical were observed to be a ff ected by solvents and a largely water containing solvent can be in favor of the deprotection reaction.
Key words:Benzoin,Caged compound,Photo-deprotection,Time-resolved infrared spectroscopy
†Part of the special issue for“the Chinese Chemical Society’s 14th National Chemical Dynamics Symposium”.
∗Authors to whom correspondence should be addressed.E-mail: kunhui@bnu.edu.cn,hongmei@iccas.ac.cn
I.INTRODUCTION
Photosensitive protecting groups,also called caged compounds,have wide applications in the spatially and temporally controlled release of biologically active compounds by exposure to light alone[1−6].Among the various protecting groups,aromatic α-keto groups have attracted great interests owing to their potential use as phototriggers for the fast and e ffi cient liberation of various biologically active stimulants[1,7−14].Especially,benzoin caged compounds have received increasing attention in this regard for their attractive photo-deprotection properties such as rapid liberation rate,relatively high quantum yield for release of leaving group and the generation of a highly fl uorescent but biologically compatible benzofuran byproduct[8, 15−17].The practical features of these compounds have led to their extensive development as photoremovable protecting groups(PRPGs)for inorganic phosphates, nucleotides and amino acid neurotransmitters,etc.[15, 18].
For the benzoin caged compounds,two di ff erent photo-deprotection reaction pathways dependent on the solvent were proposed via detecting the byproducts and intermediate species by the transient absorption spectroscopy(Scheme 1)[19,20].In CH3CN,the deprotection reaction of benzoin diethyl phosphate was found to be a rapid unimolecular process leading to elimination of the caged molecule diethyl phosphoric acid,which is concurrent with the cyclization to yield the benzofuran product.In mixed solvent with high ionizing ability,such as water or tri fl uoroethanol,the reaction leads to not only the deprotection-cyclization,but also a heterolytic triplet cleavage of the keto α-C−O bond to release the caged molecule through a branching and competing reaction.Additionally,it has been reported that the photolysis of benzoin caged compounds can also lead to homolytic cleavage of the C−C=O bond [15,16,21,22].However,although numerous studies on the photochemical reaction have been documented in the literature,knowledge is rather limited concerning the intermediate processes of homolysis reaction accompanying the deprotection reaction due to the limit of detection techniques.Thus,although the uncaging products appear to be well-determined,the underlying details of the reaction mechanisms involving the homolysis side reactions as well as the associated solvent dependence have remained unclear.
In these regards,we are motivated to investigate the photochemical processes of the benzoin caged compound,o-(2-methylbenzoyl)-DL-benzoin(MBBZ),and explore the in fl uence of water on the reaction processes under 266 nm laser irradiation by time-resolved infrared (TRIR)spectroscopy.Due to the speci fi c vibrational
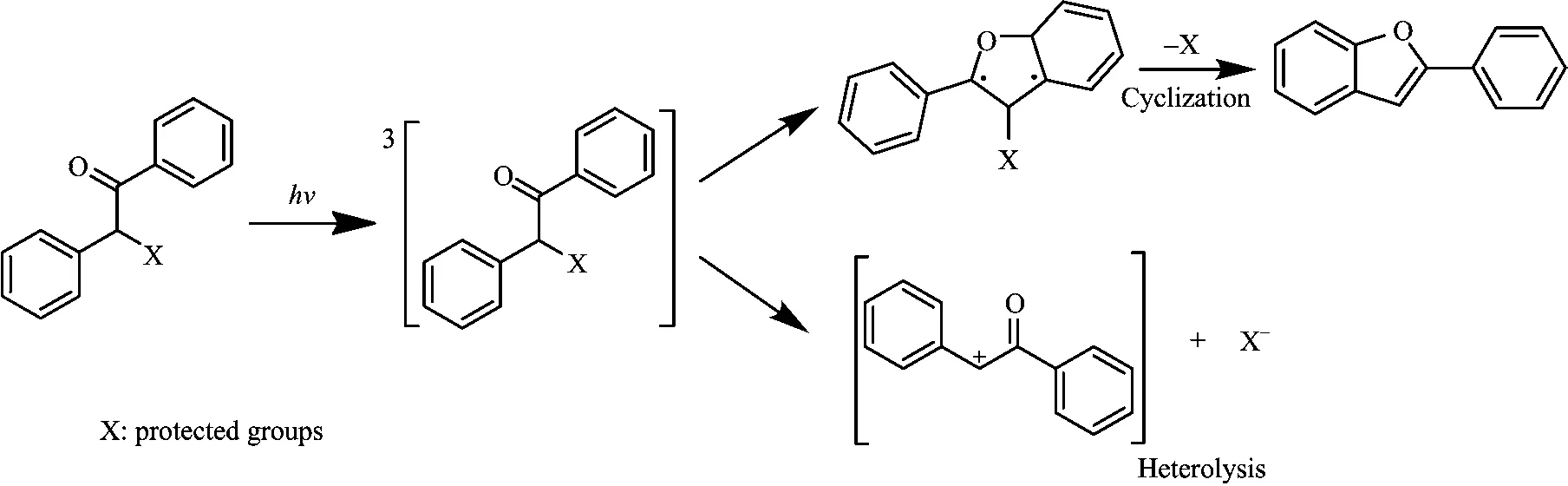
Scheme 1 Photo-deprotection reaction pathways of benzoin caged compounds. marker bands and the IR discerning capability,timeresolved infrared(TRIR)spectroscopy in the nanosecond time domain is a powerful technique,which is performed to directly probe and determine the photochemical reaction[23−25].In the TRIR spectra, we observed the 2-methylbenzoic acid from the main photo-deprotection reaction and the key radical intermediates for the photocleavage of desyl derivatives at C−C=O bond.Furthermore,the dependence of yields of uncaging product 2-methylbenzoic acid and benzoyl radical on water content indicated that the large water content in the CH3CN/D2O mixed solvent may promote the deprotection reaction and inhibit the competing homolysis of C−C=O bond.These study results may shed light on the photochemical mechanisms of the benzoin caged compounds,which can be a significant guidance in the biological applications of benzoin caged compounds.
II.EXPERIMENTAL AND COMPUTATIONAL METHODS
A.Materials
o-(2-Methylbenzoyl)-DL-benzoin(SigmaAldrich) anddeuteroxide(D2O,SigmaAldrich,>99.9%), 2-methylbenzoicacid(TCI,>98%),benzil(TCI, >99%)were used without further puri fi cation.HPLC grade acetonitrile(CH3CN)was used as solvent.
B.Time-resolved TRIR
The photochemical reaction is monitored by stepscan TRIR absorption spectroscopy[26,27].Detailed experimental procedures for TRIR absorption spectroscopy have been described in our previous work[25]. Brie fl y,the TRIR instrument(setup shown in Fig.1) comprises a Nicolet Nexus 870 step-scan FTIR spectrometer,a Continuum Surelite II Nd:YAG laser,a pulse generator(Stanford Research DG535)to initiate
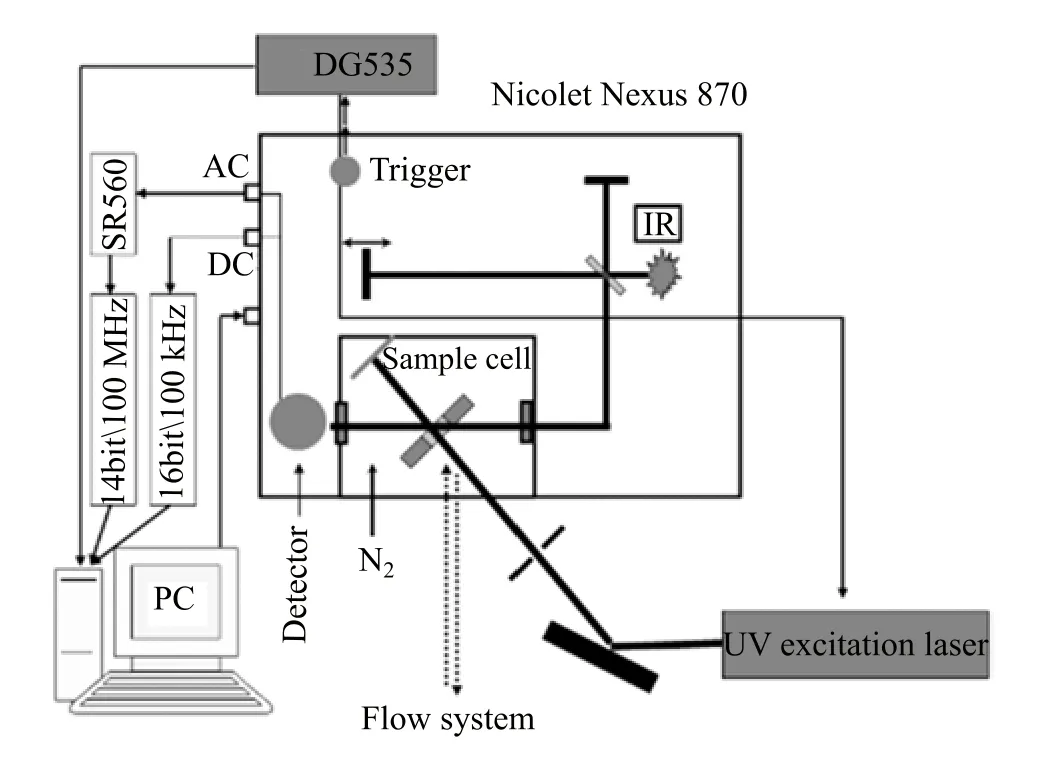
FIG.1 Schematic representation of the TRIR experimental setup.

the laser pulse and achieve synchronization of the laser with data collection,two digitizers(internal 100 kHz 16-bit digitizer and external 100 MHz 14-bit GAGE 14100 digitizer)which o ff er fast time resolution and a wide dynamic range as needed,and a personal computer to control the whole experiment.The detector used in this work is the photovoltaic MCT(0.5 mm)equipped with a fast internal preampli fi er(50 MHz).There are two outputs from the detector:output DC,corresponding to the value of the static interferogram;and output AC,corresponding to the time-resolved change of the interferogram.The AC signal is then ampli fi ed by an external preampli fi er(Stanford Research,SR560).The di ff erential absorbance spectra are calculated based on the following equation:where IDCand∆IACare the single-beam intensity spectra corresponding to static(DC)and dynamic(AC) channels.∆IACis calibrated before being used in equation because di ff erent gain is applied to the AC channel[26,27].The fourth harmonic of Nd:YAG laser (266 nm)operating at 10 Hz repetition rate was used in the experiments.The laser excitation beam was directed through an iris aperture(3 mm in diameter)and then overlapped with the infrared beam in the sample cell within the sample compartment of the FTIR spectrometer.The laser beam energy after the aperture was 2 mJ/pulse.The IR spectra were collected with a spectral resolution of 8 cm−1.A Harrick fl owing solution cell with 2 mm thick CaF2windows(pathlength,200µm)was used for the measurements.The closed fl owing system is driven by a peristaltic pump (ColeParmer Master fl ex)to refresh the sample before every laser pulse.
C.Computational methods
Theoretically,the geometries of the ground state MBBZ,possible intermediates and stable products are optimized using the hybrid density functional theory, i.e.,Becke’s three-parameter nonlocal exchange functional with the nonlocal correlation functional of Lee, Yang,Parr(B3LYP)with the 6-311+G(d,p)basis sets [28,29].Harmonic vibrational frequencies and relative intensities are therefore calculated at the same level with the optimized geometries in solvents under the polarized continuum model(PCM)[30].All of the theoretical calculations are performed with the Gaussian 09 program package[31].
III.RESULTS AND DISCUSSION
A.Identi fi cation of the reaction intermediates/products from TRIR spectra
TRIR measurements were performed to obtain the detailed(spectral and kinetic)information for photochemical reaction of MBBZ.Figure 2(b)displays the TRIR di ff erence spectra of MBBZ in CH3CN at typical delay times following 266 nm photolysis.The di ff erence spectra represent the di ff erence between the spectra obtained after photolysis and the spectrum before photolysis.The depletion of reactant gives rise to negative signals,and the formation of transient intermediates or products leads to positive bands.As shown in Fig.2(b), immediately after UV excitation,two intense bleaching bands at 1697 and 1722 cm−1were observed,which is ascribed to the depletion of ground state molecules MBBZ,as further con fi rmed by the steady-state IR absorption spectrum of MBBZ(Fig.2(a)).To aid with spectral assignment,IR frequencies and IR intensities were calculated at the B3LYP/6-311+G(d,p)level for the possible relevant species involved in the photochemical reaction,with the solvation e ff ect(CH3CN)simulated by PCM model(Table I).The calculated IR frequencies of 1714 and 1727 cm−1for MBBZ are in good agreement with the experimentally observed peak wavelength at 1697 and 1722 cm−1in the steady-state IR absorption spectrum(Fig.2(a)),which indicates that the current level of calculations can provide reliable IR frequencies for spectral assignment purpose.

FIG.2(a)Steady-state IR spectrum of 5 mmol/L MBBZ in acetonitrile.(b)Transient IR spectra of 5 mmol/L MBBZ in acetonitrile at selected time delays saturated with N2following 266 nm laser irradiation.Inset:kinetic traces curves for 1820 and 1675 cm−1.(c)Steady-state di ff erence spectrum of 5 mmol/L MBBZ in acetonitrile saturated with N2after 1 min of 266 nm laser irradiation.Inset:the longtime kinetic trace for 1735 cm−1.
In accompany with the reactant bleaching bands at1697 and 1722 cm−1,two prominent positive bands at 1735 and 1820 cm−1are formed immediately within the fi rst 0.6µs.Additionally,another positive band at 1675 cm−1is gradually formed after 0.6µs.According to the calculated vibrational frequencies(Table I), the 1820 cm−1band can be ascribed to the benzoyl radical(A1)from the homolytic cleavage of C−C=O bond,which is the only species with the vibrational frequency close to 1820 cm−1.The recombination of benzoyl radicals lead to the stable product benzil,which should correspond to the gradually formed band at 1675 cm−1in Fig.2(b).The predicted vibrational frequencies for benzil(Table I)is quite close to the observed wide peak positions of 1675 cm−1both in the transient spectra(Fig.2(b))and in the steady-state IR absorption di ff erence spectrum of N2-saturated MBBZ solution recorded after 266 nm irradiation(Fig.2(c)). Thus,the 1675 cm−1band can be ascribed to the benzil formed by the recombination of two benzoyl radicals (A1).To con fi rm this assignment,the IR spectrum of the authentic sample of benzil in CH3CN was recorded (Table I)and found to show a strong wide absorption at 1678 cm−1attributed to the antisymmetric stretching vibration and symmetrical stretching vibration of two C=O.The spectral positions of benzil match well with the observed IR di ff erence spectra(Fig.2(c))after UV irradiation which exhibits a strong wide positive band at 1675 cm−1.The small peak shift of 3 cm−1 in the IR di ff erence spectra after UV irradiation results from the partial overlapping of the positive benzil band with the negative band of MBBZ,as well as the slight ground state recovery for 1696 cm−1in the transient absorption spectra.Additionally,as shown in the inset of Fig.2(b),the decay kinetics of 1820 cm−1for benzoyl radical(A1)and the formation kinetics of 1675 cm−1 for the recombination product benzil follow almost the same time scale,providing a supplementary evidence to con fi rm the spectral assignment.
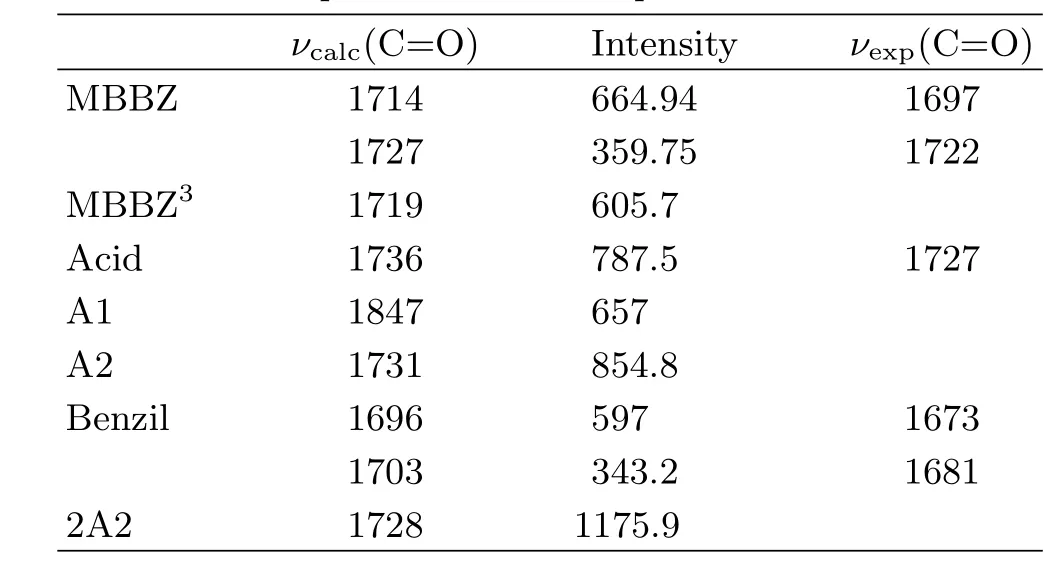
TABLE I B3LYP/6-311+G(d,p)calculated IR frequencies (in cm−1)and IR intensities(km/mol)for carbonyl stretching modes of MBBZ and relevant species in CH3CN,with the solvent e ff ect simulated by PCM model,as well as the observed IR frequencies for these species.

For the positive band at 1735 cm−1,its spectrum intensity decays to a plateau,as the kinetic trace shown in the inset of Fig.2(c),which indicates that this band should consist of two components,a transient species and a stable photoproduct respectively.In fact,the excellent agreement between the TRIR spectra and the steady-state IR di ff erence spectrum for 1732 cm−1further con fi rms that stable photoproduct contributes to the positive band at 1735 cm−1.For the transient species,as shown in the Table I,the predicted vibrational frequencies for 2-methylbenzoate benzyl radical(A2)and triplet state of MBBZ are both close to 1735 cm−1in the transient spectra.The transient 1735 cm−1band is ascribed to the A2 radical but not the triplet state because the short lifetime of 10−25 ns for triplet state,according to the previous reports[19,20],is out of the current instrument response range(100 ns).For the stable product peaked at 1732 cm−1,both 2A2 and the uncaging product 2-methylbenzoic acid can contribute to this positive band and it is hard to distinguish.To assist the assignment of the stable product at 1732 cm−1,di ff erent mixed solvents are used to measure the steadystate spectra.As shown in Fig.3,the IR characteristic marker band of the 2-methylbenzoic acid can shift from 1727 cm−1to 1700 cm−1with the increase of water concentration to 25%in D2O/CH3CN mixed solvents,while the absorption spectrum of MBBZ is almost una ff ected by the D2O.In the steady-state infrared di ff erence absorption spectra shown in Fig.3(b), the positive band of 1732 cm−1in the CH3CN solution is shifted to 1711 cm−1after the addition of 4%D2O,which is in agreement with the shift observed for 2-methylbenzoic acid,indicating that the stable species peaked at 1732 cm−1can be ascribed to the uncaging product 2-methylbenzoic acid.Additionally, the contribution of 2A2 to the 1732 cm−1can be excluded through evaluating the solvent in fl uence as detailedly discussed in the following.
B.Determination of the photochemical processes of MBBZ

FIG.3IR spectra in 4%D2O:96%CH3CN(red line), CH3CN(black line),and 25%D2O:75%CH3CN(blue line). (a)Steady-state IR spectra of 2 mmol/L 2-methylbenzoic acid.(b)Steady-state di ff erence spectra of 5 mmol/L MBBZ saturated with N2after 1 min of 266 nm laser irradiation. (c)Steady-state IR spectra of 5 mmol/L MBBZ.
From TRIR spectra,two intermediate radicals and two stable products were observed and two di ff erent channels can be elucidated.First,the observed stable product 2-methylbenzoic acid should result from the deprotection channel of MBBZ,which is the primary channel to give rise to the uncaging product.For the other stable product benzil,it may arise from the homolysis of MBBZ as postulated in the previous studies.In the current experiment,we have observed the intermediate benzoyl radical A1 and the other intermediate of the homolysis,the 2-methylbenzoate benzyl radical A2 in our TRIR spectra.In addition,the benzoyl radical recombination product benzil was observed.The direct detection of these two intermediate radicals provides explicit evidence for the existence of the homolysis of MBBZ in competition with the uncaging process of benzoin caged compounds.Nevertheless,there is no ground state recovery for the negative peak 1722 cm−1in the transient absorption spectra indicating that the recombination of A1 and A2 to form the precursor molecule can be ruled out.Moreover,upon addition of pentadiene,an e ffi cient triplet quencher,all of the transient features disappear in the TRIR spectra(spectra not shown).This means that both the above two reaction channels are associated with the triplet state of MBBZ,which is consistent with the previous reports on other benzoin caged compounds[19,20,32,33].The photochemical reaction mechanisms of MBBZ in CH3CN after 266 nm irradiation can be summarized in Scheme 2.

FIG.4 Transient IR spectra of 5 mmol/L MBBZ at selected time delays saturated with N2following 266 nm laserirradiation:(a)in4%D2O:96%CH3CN;(b)in 25%D2O:75%CH3CN.Inset:kinetic traces for 1735 cm−1 respectively.
C.Evaluation of the solvent in fl uence
The key transient intermediates identi fi ed in the TRIR spectra by their characteristic vibrational marker bands,provide direct spectral evidence to establish the side reaction of photocleavage of desyl derivatives at C−C=O bond,which is in competition with the main uncaging reaction to release 2-methylbenzoic acid.How to restrain the side reaction and promote the main reaction?We testi fi ed the solvent e ff ect and evaluated the e ff ect of water on the uncaging reaction and the homolysis reaction of C−C=O bond.TRIR absorption spectra were measured in the 4%D2O:96%CH3CN and 25%D2O:75%CH3CN respectively,as shown in Fig.4.Compared with the pure CH3CN solution,the 1735 cm−1band decays to the baseline as shown in

Scheme 2 Photochemical reaction mechanism of MBBZ in CH3CN after 266 nm irradiation. the insets of the Fig.4,indicating that this band is totally ascribed to the A2 radical intermediate and no stable product 2A2 is detected in the spectra.In the presence of 4%D2O,the stable uncaging product 2-methylbenzoic acid that was mixed in the 1735 cm−1 band should be shifted to lower frequency of 1711 cm−1, as suggested in the steady-state spectra(Fig.3).In this case,the 2-methylbenzoic acid band is buried between the two ground state bleaching bands,which makes the band intensity at 1711 cm−1increase and approach to positive(Fig.4),which is unlike the deep dips in pure CH3CN solution(Fig.3).
One advantage of the TRIR absorption spectra is that it measures simultaneously the depletion of reactant molecules with the formation of photoproducts.Thus,both the amount of photoproduct formed and reactant consumed can be quanti fi ed by normalizing the peak area of the IR absorption band with the absorption coe ffi cient.Therefore,we can investigate the in fl uence of water on the branching ratios of the deprotection reaction and homolytic cleavage of C−C=O by estimating the yields of 2-methylbenzoic acid and benzoyl radical or A2.The extinction coeffi cients of the reactant and 2-methylbenzoic acid were obtained from its steady-state FTIR spectra based on the Beer-Lambert law.By comparing the B3LYP/6-311+G(d,p)calculated IR intensities,which are proportional to their corresponding extinction coe ffi cients, the extinction coe ffi cients of benzoyl radical and A2 can be determined.The IR extinction coe ffi cients of the relevant species are listed in Table II.Because 2-methylbenzoic acid,benzoyl radical,A2,and the reactant all reach their maximum spectral intensities at 0.6µs simultaneously,both the amount of the photochemical product formation and the reactant consumption are quanti fi ed by normalizing the peak area of the IR absorption band at their maximum spectral in-

TABLE II ν(C=O)(in cm−1)and their extinction coeffi cients ε(in(mol/L)−1cm−1)of MBBZ,2-methylbenzoic acid,benzoyl radical,and A2.
aThese values are estimated according to the measured steady state IR absorption spectra.bThe extinction coe ffi cients of benzoyl radical and A2 are estimated by comparing its calculated IR intensity with that of reactant(MBBZ),based on the fact that the IR intensities are proportional to the corresponding extinction coe fi cients.
tensities(obtained by Lorentzian fi tting as shown in Fig.5)with the absorption coe ffi cient listed in Table II. Furthermore,dividing the amount of photoproduct formation by reactant consumption gives the absolute yields.The yields for the two photochemical products 2-methylbenzoic acid and benzoyl radical are 46.7%and 38.5%in the pure CH3CN and 47.2%and 37.6%in the 4%D2O:96%CH3CN.The similar yields indicate that little amount of water has almost no e ff ect on the photochemical processes.Moreover,it is con fi rmed that there is neither contribution of 2A2 for the 1732 cm−1 in the pure CH3CN nor that in the 4%D2O:96%CH3CN. However,in the 25%D2O:75%CH3CN,the yield of the uncaging 2-methylbenzoic acid grows to 55.0%and the yield of benzoyl radical reduces to 31.9%.It is demonstrated that large amount of water can promote thephoto-deprotection reaction.This is probably due to the good ionizing and dissociating capability makes water an ideal solvent for the e ffi ciently heterolytic triplet cleavage of the keto α-C=O bond to occur and thus facilitates the uncaging reaction.Additionally,according to the fi tting results shown in Fig.5,the yields of A1 and A2 are 38.5%and 39.2%in the pure CH3CN, 37.6%and 41.2%in the 4%D2O:96%CH3CN,31.9%and 33.9%in the 25%D2O:75%CH3CN,respectively,which demonstrates that the yields of A1 and A2 are almost identical considering the error range in the di ff erent solvent system,further con fi rming the spectral assignment of A1 and A2 from the homolysis of MBBZ.

FIG.5 Experimental TRIR di ff erence spectra(black dots) of 5 mmol/L MBBZ in(a)pure CH3CN solution,(b)D2O and CH3CN mixed solution(4%D2O:96%CH3CN),(c)D2O and CH3CN mixed solution(25%D2O:75%CH3CN)at 0.6µs after 266 nm excitation with the fi tted spectrum(red line), composed of individual Lorentzian bands shown with green lines(the ground-state MBBZ bleaching bands),dark yellow lines(homolysis radical positive bands),and blue lines(the 2-methylbenzoic acid positive band).
Overall,our experiments provide direct spectral evidence to the homolytic cleavage reaction of C−C=O bond,which is in competition with the release of 2-methylbenzoic acid after 266 nm laser irradiation MBBZ in CH3CN solution.Moreover,the product yields of 2-methylbenzoic acid and benzoyl radical were observed to be a ff ected by the presence of large amount of water,indicating that deprotection reaction is favorable in the water containing solvent.
IV.CONCLUSION
In this work,we have performed time-resolved infrared spectroscopy studies of the photochemical reaction of benzoin caged compound o-(2-methylbenzoyl)-DL-benzoin.By taking advantage of the speci fi c vibrational marker bands and the IR discerning capability, we have identi fi ed and distinguished the uncaging product 2-methylbenzoic acid and two key intermediate radicals of C−C=O bond homolytic cleavage in the transient infrared spectra.Two photochemical channels, deprotection channel to form 2-methylbenzoic acid and the homolysis of MBBZ to generate benzil,are therefore determined.The key transient intermediates identi fi ed in the TRIR spectra provide direct spectral evidence to establish the side reaction of photocleavage of desyl derivatives at C−C=O bond.Moreover,the dependence of the product yields on the water content shows that very small amount of water has no e ff ect on the reaction processes,while a largely water containing solvent can be in favor of the deprotection reaction.These results provide mechanistic insights to guide the biological applications of benzoin caged compounds,especially in reducing the side reactions of caged compounds.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.21333012 and No.21425313)and the National Basic Research Program of China(No.2013CB834602).
[1]R.S.Givens and L.W.Kueper,Chem.Rev.93,55 (1993).
[2]G.P.Hess,Biochemistry 32,989(1993).
[3]G.M.Staub,J.B.Gloer,D.T.Wicklow,and P.F. Dowd,J.Am.Chem.Soc.114,1015(1992).
[4]G.C.R.Ellis-Davies,Nat.Methods 4,619(2007).
[5]G.Mayer and A.Heckel,Angew.Chem.Int.Ed.45, 4900(2006).
[6]M.Lukeman and J.C.Scaiano,J.Am.Chem.Soc.127, 7698(2005).
[7]Y.Q.Yu,L.D.Wu,X.R.Zou,X.J.Dai,K.H.Liu, and H.M.Su,J.Phys.Chem.A 117,7767(2013).
[8]A.P.Pelliccioli and J.Wirz,Photochem.Photobiol. Sci.1,441(2002).
[9]K.Curley and D.S.Lawrence,J.Am.Chem.Soc.120, 8573(1998).
[10]C.Ma,W.M.Kwok,W.S.Chan,P.Zuo,J.T.Wai Kan,P.H.Toy,and D.L.Phillips,J.Am.Chem.Soc. 127,1463(2005).
[11]M.Zabadal,A.P.Pelliccioli,P.Kl´an,and J.Wirz,J. Phys.Chem.A 105,10329(2001).
[12]C.S.Ma,W.M.Kwok,W.S.Chan,Y.Du,J.T.W. Kan,P.H.Toy,and D.L.Phillips,J.Am.Chem.Soc. 128,2558(2006).
[13]R.S.Givens,J.F.W.Weber,P.G.Conrad,G.Orosz, S.L.Donahue,and S.A.Thayer,J.Am.Chem.Soc. 122,2687(2000).
[14]M.A.Hangarter,A.H¨ormann,Y.Kamdzhilov,and J. Wirz,Photochem.Photobiol.Sci.2,524(2003).
[15]K.R.Gee,L.W.Kueper,J.Barnes,G.Dudley,and R. S.Givens,J.Org.Chem.61,1228(1996).
[16]J.C.Sheehan and R.M.Wilson,J.Am.Chem.Soc. 86,5277(1964).
[17]R.S.Givens and B.Matuszewski,J.Am.Chem.Soc. 106,6860(1984).
[18]R.S.Givens,P.S.Athey,L.W.Kueper,B.Matuszewski,and J.Y.Xue,J.Am.Chem.Soc.114,8708 (1992).
[19]C.Ma,Y.Du,W.M.Kwok,and D.L.Phillips,Chem. Eur.J.13,2290(2007).
[20]C.S.Rajesh,R.S.Givens,and J.Wirz,J.Am.Chem. Soc.122,611(2000).
[21]J.F.Cameron,C.G.Willson,and J.M.J.Fr´echet,J. Am.Chem.Soc.118,12925(1996).
[22]R.Kaliappan,L.S.Kaanumalle,and V.Ramamurthy, Chem.Commun.4056(2005).
[23]X.R.Zou,X.J.Dai,K.H.Liu,H.M.Zhao,D.Song, and H.M.Su,J.Phys.Chem.B 118,5864(2014).
[24]C.F.Yang,Y.Q.Yu,K.H.Liu,D.Song,L.D.Wu, and H.M.Su,J.Phys.Chem.A 115,5335(2011).
[25]W.Q.Wu,K.H.Liu,C.F.Yang,H.M.Zhao,H. Wang,Y.Q.Yu,and H.M.Su,J.Phys.Chem.A 113, 13892(2009).
[26]W.Uhmann,A.Becker,C.Taran,and F.Siebert,Appl. Spectrosc.45,390(1991).
[27]D.L.Drapcho,R.Curbelo,E.Y.Jiang,R.A.Crocombe,and W.J.McCarthy,Appl.Spectrosc.51,453 (1997).
[28]C.Lee,W.Yang,and R.G.Parr,Phys.Rev.B 37,785 (1988).
[29]A.D.Becke,J.Chem.Phys.98,5648(1993).
[30]B.Mennucci and J.Tomasi,J.Chem.Phys.106,5151 (1997).
[31]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E. Scuseria,M.A.Robb,J.R.Cheeseman,G.Scalmani, V.Barone,B.Mennucci,G.A.Petersson,H.Nakatsuji,M.Caricato,X.Li,H.P.Hratchian,A.F.Izmaylov,J.Bloino,G.Zheng,J.L.Sonnenberg,M. Hada,M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa, M.Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai, T.Vreven,J.A.Montgomery Jr.,J.E.Peralta,F. Ogliaro,M.Bearpark,J.J.Heyd,E.Brothers,K.N. Kudin,V.N.Staroverov,R.Kobayashi,J.Normand, K.Raghavachari,A.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,J.M.Millam,M. Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo, J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev, A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochterski, R.L.Martin,K.Morokuma,V.G.Zakrzewski,G.A. Voth,P.Salvador,J.J.Dannenberg,S.Dapprich,A. D.Daniels,¨O.Farkas,J.B.Foresman,J.V.Ortiz,J. Cioslowski,and D.J.Fox,Gaussian 09,Revision A.1, Wallingford CT:Gaussian,Inc.,(2009).
[32]M.Lipson and N.J.Turro,J.Photochem.Photobiol. A 99,93(1996).
[33]F.D.Lewis,R.T.Lauterbach,H.G.Heine,W.Hartmann,and H.Rudolph,J.Am.Chem.Soc.97,1519 (1975).
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- ARTICLE E ffi cient Separation of Ar and Kr from Environmental Samples for Trace Radioactive Noble Gas Detection†
- REVIEW Polarization Dependent Time-Resolved Infrared Spectroscopy and Its Applications†
- ARTICLE Reactions of Group V Metal Atoms with Hydrogen Sul fi de:Argon Matrix Infrared Spectra and Theoretical Calculations†
- ARTICLE Structural Dynamics of Phenyl Azide in Light-Absorbing Excited States: Resonance Raman and Quantum Mechanical Calculation Study†
- ARTICLE Structural and Infrared Spectroscopic Study on Solvation of Acetylene by Protonated Water Molecules†
- ARTICLE Excited-State Proton Transfer and Decay in Hydrogen-Bonded Oxazole System:MS-CASPT2//CASSCF Study†