ARTICLE Infrared Photodisssociation Spectroscopy of Boron Carbonyl Cation Complexes†
2016-04-08JiayeJinGuanjunWangMingfeiZhouCollaborativeInnovationCenterofChemistryforEnergyMaterialsDepartmentofChemistryShanghaiKeyLaboratoryofMolecularCatalystsandInnovativeMaterialsFudanUniversityShanghai200433ChinaDatedReceived
Jia-ye Jin,Guan-jun Wang,Ming-fei Zhou∗Collaborative Innovation Center of Chemistry for Energy Materials,Department of Chemistry,Shanghai Key Laboratory of Molecular Catalysts and Innovative Materials,Fudan University,Shanghai 200433, China(Dated:Received on November 25,2015;Accepted on December 8,2015)
ARTICLE Infrared Photodisssociation Spectroscopy of Boron Carbonyl Cation Complexes†
Jia-ye Jin,Guan-jun Wang,Ming-fei Zhou∗
Collaborative Innovation Center of Chemistry for Energy Materials,Department of Chemistry,Shanghai Key Laboratory of Molecular Catalysts and Innovative Materials,Fudan University,Shanghai 200433, China
(Dated:Received on November 25,2015;Accepted on December 8,2015)
The boron carbonyl cation complexes B(CO)3+,B(CO)4+and B2(CO)4+are studied by infrared photodissociation spectroscopy and theoretical calculations.The B(CO)4+ions are characterized to be very weakly bound complexes involving a B(CO)3+core ion,which is predicted to have a planar D3hstructure with the central boron retaining the most favorable 8-electron con fi guration.The B2(CO)4+cation is determined to have a planar D2hstructure involving a B−B one and half bond.The analysis of the B-CO interactions with the EDANOCV method indicates that the OC→B σ donation is stronger than the B→CO π back donation in both ions.
Key words:Boron carbonyl,Donor-acceptor bonding,Infrared photodissociation spectroscopy,Theoretical calculations
†Part of the special issue for“the Chinese Chemical Society’s 14th National Chemical Dynamics Symposium”.
∗Author to whom correspondence should be addressed.E-mail: mfzhou@fudan.edu.cn
I.INTRODUCTION
Carbon monoxide is one of the most important ligand in inorganic and organometallic chemistry[1].It can bind to a host of neutral and charged transition metal centers in forming diverse metal carbonyl complexes,in which carbon monoxide serves either as a two-electron donor in an end-on coordinated fashion or as a four-electron or even six-electron donor in the bridge bonded modes[2−11].Carbon monoxide can also coordinate with some main-group elements in forming main group carbonyl complexes[12,13].Homoleptic mono-and dicarbonyl complexes of main group elements with end-on bonded carbonyl ligands have been prepared and spectroscopically characterized either in low-temperature noble gas matrices or in the gas phase[12−38].These carbonyl complexes are usually not stable at ambient conditions with the exception of[C(CO)2]and[N(CO)2]+which have been crystallographically characterized[36−38].
In the case of boron,the electron de fi cient boron species are able to coordinate one CO ligand in forming the closed-shell carbonyl borane H3BCO and related derivatives,which are well-known stable carbonyl compounds[39−43].Boron carbonyl species such as BCO,B(CO)2,B(CO)2−,OCBBCO,BBCO and B4(CO)2were identi fi ed as products from the reactions of thermal-or laser-evaporated boron atoms with CO in solid argon[44−50].The OCBBCO molecule was characterized to be a boron-boron triple bonded species[48].Both BBCO and B4(CO)2are σ-π diradicals[49,50].Bonding analysis suggests that the linear closed-shell B(CO)2−anion should be considered as a donor-acceptor bonding complex rather than a cumulene O=C=B(−)=C=O molecule with electron sharing bonding[47].Very recently,a rare example of a boron dicarbonyl complex[(RB)(CO)2](R being a bulky aryl group)with two terminal carbonyl ligands which is stable under ambient conditions has been reported[51].The chemical behavior shows typical features of carbonyl complexes which are known from transition metal carbonyls.Boron carbonyl cation complexes are not known so far.Here we report a combined infrared photodissociation spectroscopy and theoretical study on boron carbonyl cation complexes B(CO)3+and B2(CO)4+in the gas phase.
II.EXPERIMENTAL AND THEORETICAL METHODS
The boron carbonyl cation complexes are generated in the gas phase using a pulsed laser vaporization/supersonic expansion ion source as described previously[52,53].Bulk targets compressed from isotopically-enriched10B and11B powders were used. The ions are produced from the laser vaporization process in expansions of helium seeded with 2%−5% CO using a pulsed valve(General Valve,Series 9)at 0.5−1.0 MPa backing pressure.After free expansion and cooling,the cations are skimmed into a second chamber where they are pulse-extracted into a Wiley-McLaren type time-of- fl ight mass spectrometer.The cations of interest are mass selected and decelerated into the extraction region of a second collinear timeof- fl ight mass spectrometer,where they are dissociated by a tunable IR laser.The tunable IR laser used is generated by a KTP/KTA//AgGaSe2optical parametric oscillator/ampli fi er system(OPO/OPA,Laser Vision)pumped by a Continuum Surelite EX Nd:YAG laser,producing about 1.0−2.5 mJ/pulse in the range of 1800−2400 cm−1.The ion density is too low for infrared absorption spectroscopy,thus,the infrared photodissociation spectroscopy is employed to record the vibrational spectra.Resonant absorption leads to fragmentation of the ion complex.The infrared photodissociation spectrum is obtained by monitoring the yield of the fragment ion as a function of the dissociation IR laser wavelength and normalizing to parent ion signal.
The geometry optimizations have been carried out without symmetry constraints at the B3LYP level using the aug-cc-pVTZ basis sets[54−56].The harmonic vibrational frequencies were calculated with analytic second derivatives.These calculations were performed using the Gaussian 09 program[57].The gradient corrected BP86 functional in conjunction with uncontracted Slater-type orbitals(STOs)as basis functions was used for the bonding analyses[58−60].The latter basis sets for all elements have triple-ξ quality augmented by one set of polarization functions(ADF-basis set TZP).An auxiliary set of s,p,d,f,and g STOs was used to fi t the molecular densities and to represent the Coulomb and exchange potentials accurately in each SCF cycle.The BP86/TZP calculations were performed using the B3LYP/aug-cc-pVTZ optimized geometries with the program package ADF2014.10[61].
III.RESULTS AND DISCUSSION
The mass spectrum of boron carbonyl cation complexes in the m/z range of 60−150 from the laser evaporation of a10B-enriched target in expansions of helium gas seeded with 5%CO is shown in Fig.1(a).Although the mass spectra depend strongly on the parameters of the ion source such as vaporization laser power,He and CO stagnation pressures and timing,the peaks corresponding to10B(CO)3+(m/z=94)and10B2(CO)4+(m/z=132)are always the most intense peaks,suggesting that these cations are formed preferentially with high stability.The mass spectrum from the experiments with the11B-enriched target is shown in Fig.1(b).The most intense peaks shifted to m/z=95 and 134,corresponding to the11B(CO)3+and11B2(CO)4+ions,respectively.
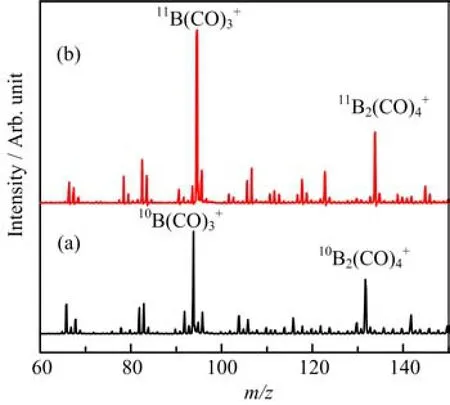
FIG.1 Mass spectra of the boron carbonyl cation complexes formed by pulsed laser vaporization of a target in an expansion of helium doped with carbon monoxide.(a)10B and (b)11B.
The B(CO)3+cations are mass-selected and subjected to infrared photodissociation.It is found that the B(CO)3+cations dissociate via losing a CO ligand when excited with infrared light in the 2140−2150 cm−1frequency region but the dissociation e ffi ciency(less than 0.5%)is too low to achieve an e ff ective spectrum. This suggests that the B(CO)3+cations are very stable species with quite high CO binding energy as expected,as it satis fi es the octet rule.In contrast to B(CO)3+,the B(CO)4+cation complexes are able to dissociate via loss of a CO ligand with very high effi ciency(>50%),indicating that B(CO)4+is a very weakly bound complex.This con fi rms our expectation that B(CO)3+is a fully coordinated ion and the fourth CO in B(CO)4+is a weakly bound external carbonyl ligand.Therefore,the B(CO)4+cation can be regarded as a CO“tagged”cation complex involving a B(CO)3+core ion.The infrared photodissociation spectrum of B(CO)4+represents the spectrum of the B(CO)3+core ion that is weakly perturbed by the tagged CO ligand. The tagging e ff ect is expected to change the position of the B(CO)3+band only slightly as discussed previously[62−64].The infrared photodissociation spectra of11B(CO)4+and10B(CO)4+in the C−O stretching frequency region are shown in Fig.2.The spectrum of10B(CO)4+(Fig.2(a))exhibits a very strong band centered at 2145 cm−1along with a weak band at 2178 cm−1.The 2145 cm−1band is just 2 cm−1blueshifted from the frequency of gas phase carbon monoxide(2143 cm−1).This band can be attributed to the antisymmetric CO stretching vibrations of the10B(CO)3+core ion.The much weak band at 2178 cm−1is assigned to the CO stretching vibration of the weakly tagged CO ligand,consistent with previous observations for other weakly bound metal ion carbonyls[64−66].The same bands were also observed in the spectrum of11B(CO)4+as shown in Fig.2(b).The band positions are essentially the same indicating that the corresponding vibrational modes are pure CO stretching vibrations with negligible boron involvement.Besides 2145 and 2178 cm−1bands, additional weak bands at 2214 and 2259 cm−1were observed in the spectrum of11B(CO)4+.Both bands are located in the frequency range suitable for the symmet-ric CO stretching vibrations.We tentatively assign the 2259 cm−1band to the symmetric stretching vibration of thecore ion and the 2214 cm−1band to a combination or an overtone level that is in Fermi resonance with the symmetric CO stretching fundamental.When11B is substituted by10B,the two levels are no long close enough to show Fermi resonance,therefore,both bands were not observed in the spectrum ofcation is able to dissociate via loss of a CO ligand under focused IR laser irradiation.The parent ions can be depleted by about 5%at the laser pulse energy of 1.4 mJ/pulse at 2108 cm−1.The infrared photodissociation spectrum ois shown in Fig.3.The spectrum exhibits two bands centered at 2108 and 2152 cm−1.
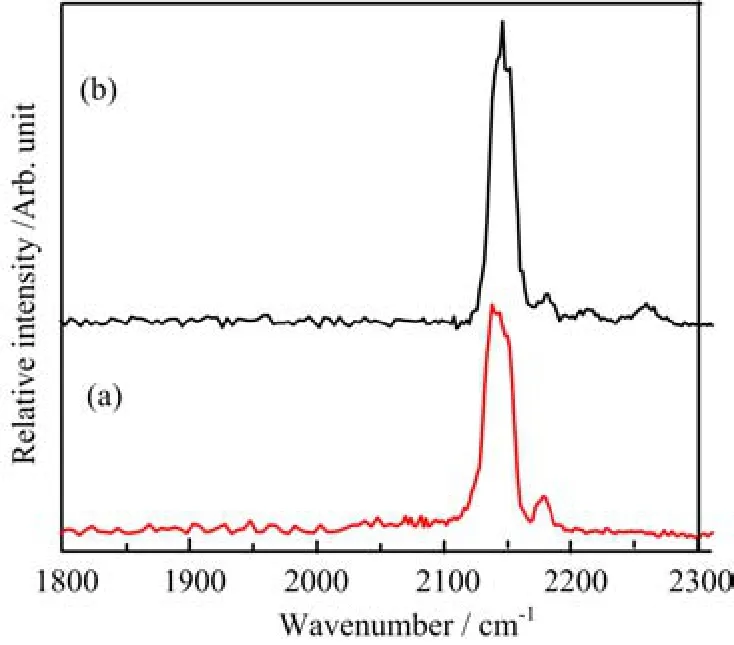
FIG.2 The experimental infrared photodissociation spectra of the(a)10B(CO)4+and(b)11B(CO)4+cation complexes in the CO stretching frequency region.
We carried out high-level calculations in order to validate the identify of the cations and to analyze their electronic structures.The theoretically predicted geometries of Bare shown in Fig.4.The B(CO)3+cation has a1A′1ground state with planar D3hsymmetry.The calculations indicate that the CO bond distances in)is slightly shorter th˚anthat of free CO calculated at the same level(1.128A).
As shown in Fig.5,the highest doubly occupied molecular orbital(HOMO,a′2′)is primarily a central B2p orbital,which comprises signi fi cant B2p to CO 2π∗back bonding.Therefore,the1A′1ground statecation correlates to an electronic excited singlet state B+with the associated valence electronic con fi guration of 2s0,2p(σ)0,2p(π)2,2p(π′)0.The11B(CO)3+cation with D3hsymmetry has only one IR active antisymmetric CO stretching mode calculated at 2223 cm−1,which is doubly degenerate.This mode foris predicted at 2224 cm−1.The symmetric stretching mode is predicted at 2317 cm−1which is IR inactive.Consistent with the experimental observations,the Bcation was predicted to be a weakly bound complex as the predicted B−CO distance of the fourth CO is quite large with the geometry of the B(CO)3+core ion being essentially the same as the free cation.Due to symmetry reduction by CO coordination,the double degeneracy of the antisymmetric CO stretching mode of B(CO)3+is lifted,and the E mode splits into two distinct modes.Calculations at the B3LYP level show very small mode split of 6 cm−1,which cannot be wellresolved experimentally.
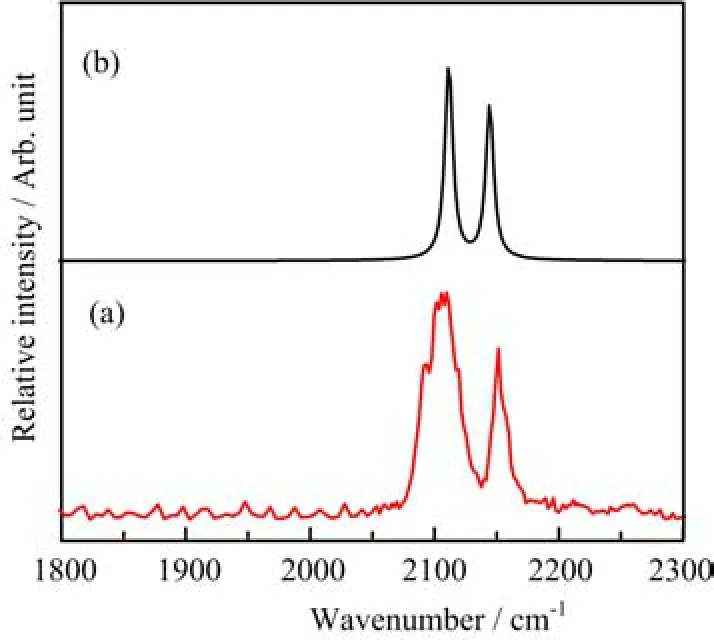
FIG.3ExperimentalandsimulatedIRspectraof
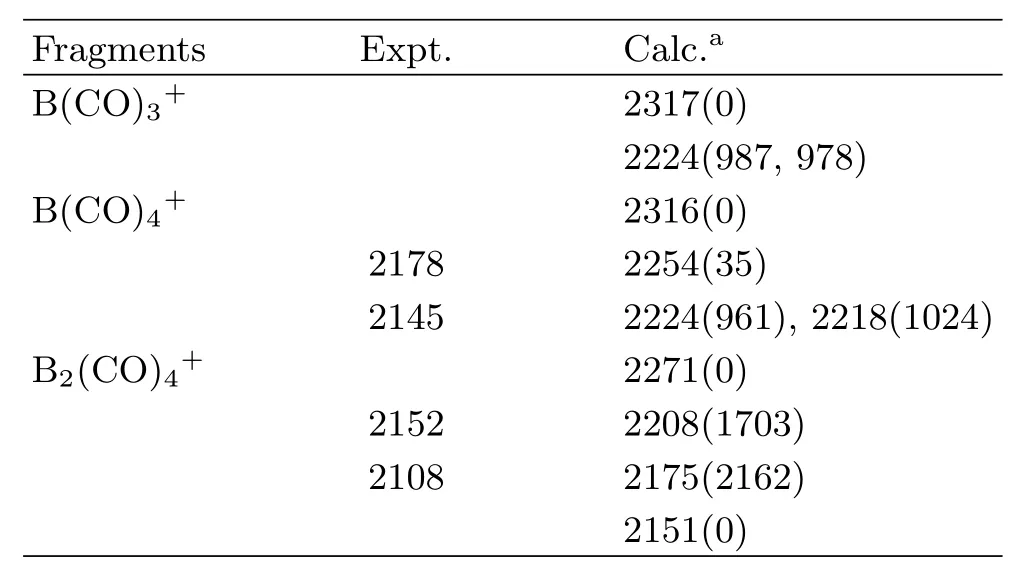
TABLE I Observed and calculated(harmonic,unscaled) CO stretching frequencies(cm−1)of the10B(CO)4+and10B2(CO)4+ions.
aThe intensities are listed in parentheses in km/mol.molecular orbital pictures shown in Fig.5 clearly indicate that the singly occupied(SOMO)b2gMO is a B−B π antibonding orbital.The highest doubly occupied b3uorbital(HOMO)is a B−B π bonding orbital.Both the b2gand b3uorbitals comprise substantial B2+to CO π∗back-donation.Both orbitals are highly delocalized involving two B and four C centers.The doubly occupied agmolecular orbital(HOMO-1)is a B−B σ bonding orbital.The ground statecation complex can thus be viewed as the interaction of aexcited-state B2+and four CO’s,which involves one B−B σ bond and a half B−B π bond.
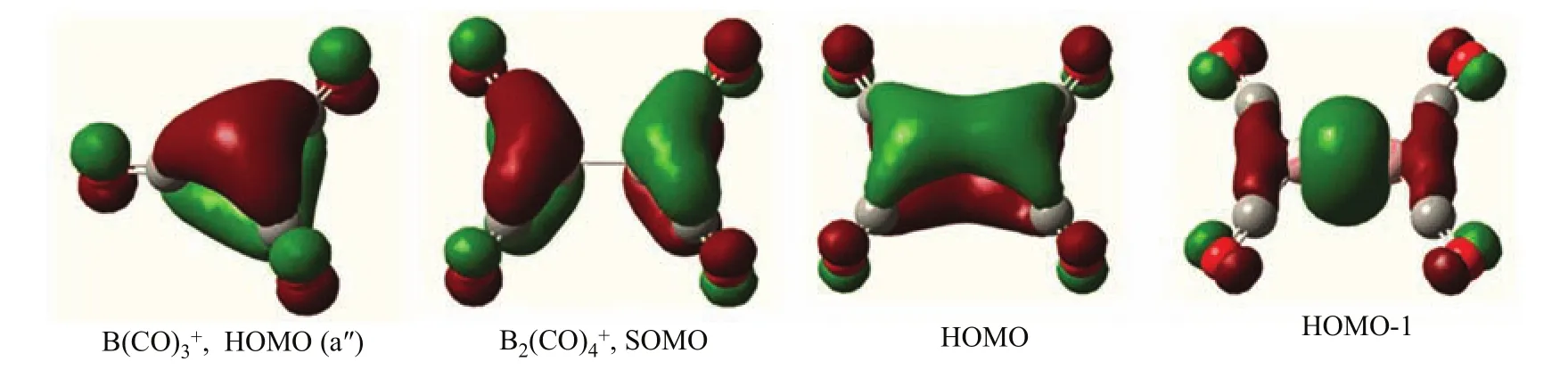
FIG.5 Molecular orbital pictures of the highest doubly occupied(HOMO)of B(CO)3+and the singly occupied(SOMO) and highest doubly occupied orbitals(HOMO and HOMO-1)of B2(CO)4+.
Thebonddissociationenergiesof,andare calculated.At the B3LYP level,the BDE ofis 79.5 kcal/mol with respect to the dissociation limit+CO or 60.7 kcal/mol with respect to the ground state reactants:.The dissociation energy of the tagged CO inis calculated to be only 2.9 kcal/mol.The binding energy of B2(CO)4+is 60.4 kcal/mol with respect to the dissociation limit B2+CO or 39.2 kcal/mol for the dissociation into the ground stateand CO.
We analyze the nature of the donor-acceptor interactions in Bwith the EDA(energy decomposition analysis)in conjunction with the NOCV(natural orbitals for chemical valence)method [68],which gives a detailed insight into the bonding situation.The numerical results of the OC−B interactions in B(CO)at the BP86/TZP level are listed in Table II.The data show that both species havevery similar orbital interaction energy(∆Eorb).Further inspection of the orbital components of∆Eorbreveals that both species have very similar CO→B σ donation interaction,which is much stronger than the B→CO π back donation interactions.There is a large di ff erence in the strength of the B→CO π back donation between the two complexes.The π contribution of∆Eorb(π⊥)+ ∆ Eorb(π‖)in B2(CO)4+is much larger than that in B(CO)3+.Figure 6 displays the deformation densities ∆ρ(σ)and∆ρ(π)which are connected to the σ donation and π backdonation in B(CO)The direction of the charge fl ow is indicated by the colors red→blue.The shape of∆ρ(σ)clearly indicates that the charge fl ow comes mainly from the lone-pairelectrons at carbon to the boron atom.While the π backdonation leads to charge accumulation mainly at the carbon atom of CO.

TABLE II EDA-NOCV results of the chemical bonding in OC-B(CO)2+and OC-B2(CO)3+at BP86/TZP.Energy values are given in kcal/mol.
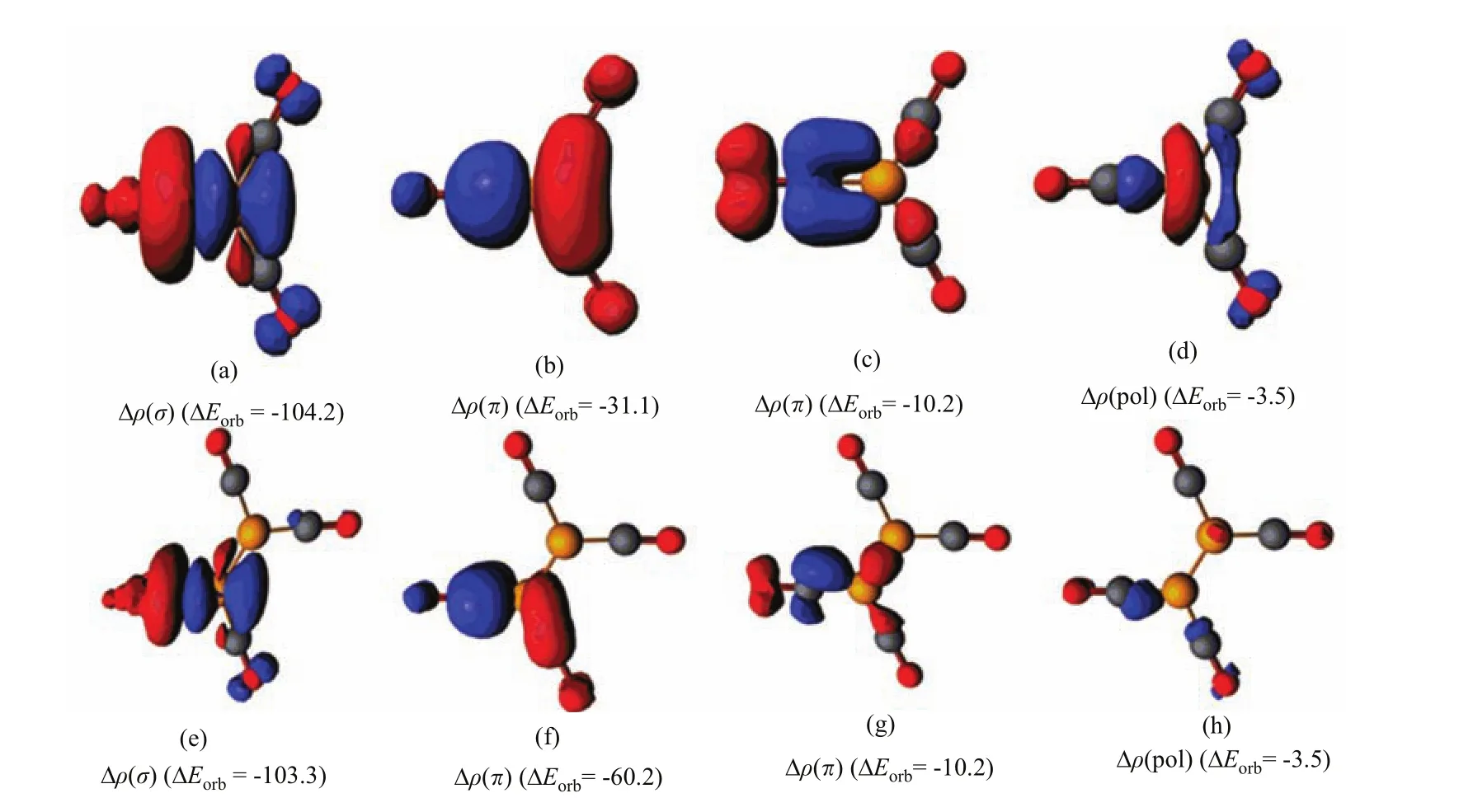
FIG.6 Plot of deformation densities∆ρ of the pairwise orbital interactions and the associated interaction energies∆Eorb(in kcal/mol)between CO and B
IV.CONCLUSION
Boron carbonyl cation complexes are produced via a laser vaporization supersonic ion source in the gas phase.The cations of interest are each mass-selected and their infrared spectra are measured via infrared photodissociation spectroscopy in the carbonyl stretching frequency region.Density functional calculations have been performed and the calculated vibrational spectra are compared to the experimental data to identify the gas-phase structures of the ions.The B(CO)3+and B2(CO)4+cations are the most intense peaks in the mass spectrum.The B(CO)3+ion is too strongly bound to achieve an e ff ective IR spectrum.In contrast, the B(CO)4+ion dissociates very e ffi ciently under IR irradiation.It is characterized to be a very weakly bound complex involving a B(CO)3+core ion,which is predicted to have a planar D3hstructure with the central boron retaining the most favorable 8-electron con fi guration.The B2(CO)4+cation is determined to have a planar D2hstructure involving a B−B bond.Both the B(CO)3+and B2(CO)4+ions have slightly red-or blue-shifted CO stretching frequencies with respect to free CO.The analysis of the B-CO interactions with the EDA-NOCV method indicates that the OC→B σ donation is stronger than the B→CO π back donation.
V.ACKNOWLEDGMENTS
The work was supported by the Ministry of Science and Technology of China(No.2013CB834603)and the National Natural Science Foundation of China (No.21173053 and No.21433005).
[1]F.A.Cotton,G.Wilkinson,C.A.Murillo,and M. Bochmann,Advanced Inorganic Chemistry,6th Edn., New York:John Wiley,(1999).
[2]G.Frenking and N.Fr¨ohlich,Chem.Rev.100,717 (2000).
[3]M.F.Zhou,L.Andrews,and C.W.Bauschlicher Jr., Chem.Rev.101,1931(2001).
[4]F.A.Cotton,B.A.Frenz,and L.Kruczynski,J.Am. Chem.Soc.95,951(1973).
[5]M.Manassero,M.Sansoni,and G.Longoni,J.Chem. Soc.Chem.Commun.919(1976).
[6]R.Colton and M.J.McCormick,Coord.Chem.Rev. 31,1(1980).
[7]L.Jiang and Q.Xu,J.Am.Chem.Soc.127,42(2005).
[8]X.J.Zhou,J.M.Cui,Z.H.Li,G.J.Wang,Z.P.Liu, and M.F.Zhou,J.Phys.Chem.A 117,1514(2013).
[9]J.H.Osborne,A.L.Rheingold,and W.C.Trogler,J. Am.Chem.Soc.107,6292(1985).
[10]X.J.Zhou,J.M.Cui,Z.H.Li,G.J.Wang,and M.F. Zhou,J.Phys.Chem.A 116,12349(2012).
[11]W.A.Herrmann,H.Biersack,M.L.Ziegler,K.Weidenhammer,R.Siegel,and D.Rehder,J.Am.Chem. Soc.103,1692(1981).
[12]A.J.Bridgeman,Inorg.Chim.Acta.321,27(2001).
[13]H.J.Himmel,A.J.Downs,and T.M.Greene,Chem. Rev.102,4191(2002).
[14]L.Andrews,T.J.Tague,and G.P.Kushto,Inorg. Chem.34,2952(1995).
[15]P.H.Kasai and P.M.Jones,J.Am.Chem.Soc.106, 8018(1984).
[16]J.H.B.Chenier,C.A.Hampson,J.A.Howard,B. Mile,and R.Sutcli ff e,J.Phys.Chem.90,1524(1986).
[17]C.Xu,L.Manceron,and J.P.Perchard,J.Chem.Soc., Faraday Trans.89,1291(1993).
[18]Q.Y.Kong,M.H.Chen,J.Dong,Z.H.Li,K.N.Fan, and M.F.Zhou,J.Phys.Chem.A 106,11709(2002). [19]L.N.Zhang,J.Dong,M.F.Zhou,and Q.Z.Qin,J. Chem.Phys.113,10169(2000).
[20]P.H.Kasai and P.M.Jones,J.Phys.Chem.89,2019 (1985).
[21]J.A.Howard,R.Sutcli ff e,C.A.Hampson,and B.Mile, J.Phys.Chem.90,4268(1986).
[22]H.J.Himmel,A.J.Downs,J.C.Greene,and T.M. Greene,J.Phys.Chem.A 104,3642(2000).
[23]W.G.Hatton,N.P.Hacker,and P.H.Kasai,J.Phys. Chem.93,1328(1989).
[24]R.R.Lembke,R.F.Ferrante,and W.Weltner,J.Am. Chem.Soc.99,416(1977).
[25]M.F.Zhou,L.Jiang,and Q.Xu,J.Chem.Phys.121, 10474(2004).
[26]A.Feltrin,S.N.Cesaro,and F.Ramondo,Vib.Spectrosc.10,139(1996).
[27]M.F.Zhou,L.Jiang,and Q.Xu,J.Phys.Chem.A 109,3325(2005).
[28]A.Bos,J.Chem.Soc.Chem.Commun.1,26(1972).
[29]L.N.Zhang,J.Dong,and M.F.Zhou,J.Chem.Phys. 113,8700(2000).
[30]L.Jiang and Q.Xu,Bull.J.Chem.Soc.Jpn.79,857 (2006).
[31]L.Jiang and Q.Xu,J.Chem.Phys.122,034505(2005). [32]L.N.Zhang,J.Dong,and M.F.Zhou,Chem.Phys. Lett.335,334(2001).
[33]A.J.Bridgeman,N.Harris,and N.A.Young,Chem. Commun.14,1241(2000).
[34]T.Liang,S.D.Flynn,A.M.Morrison,and G.E.Douberly,J.Phys.Chem.A 115,7437(2011).
[35]A.D.Brathwaite and M.A.Duncan,J.Phys.Chem. A 116,1375(2012).
[36]A.Ellern,T.Drews,and L.Seppelt,Z.Anorg.Allg. Chem.627,73(2001).
[37]R.Tonner and G.Frenking,Chem.Eur.J.14,3260 (2008).
[38]I.Bernhardi,T.Drews,and K.Seppelt,Angew.Chem. Int.Ed.38,2232(1999).
[39]A.B.Burg and H.I.Schlesinger,J.Am.Chem.Soc. 59,780(1937).
[40]A.Terheiden,E.Bernhardt,H.Willner,and F.Aubke, Angew.Chem.Int.Ed.41,799(2002).
[41]M.Finze,E.Bernhardt,A.Terheiden,M.Berkei,H. Willner,D.Christen,H.Oberhammer,and F.Aubke, J.Am.Chem.Soc.124,15385(2002).
[42]M.Gerken,G.Pawelke,E.Bernhardt,and H.Willner, Chem.Eur.J.16,7527(2010).
[43]A.Fukazawa,J.L.Dutton,C.Fan,L.G.Mercier,A.Y. Houghton,Q.Wu,W.E.Piers,and M.Parvez,Chem. Sci.3,1814(2012).
[44]Y.M.Hamrick,R.J.V.Zee,J.T.Godbout,W.Weltner,W.J.Lauderdale,J.F.Stanton,and R.J.Bartlett, J.Phys.Chem.95,2840(1991).
[45]T.R.Burkholder and L.Andrews,J.Phys.Chem.96, 10195(1992).
[46]M.F.Zhou,N.Tsumori,L.Andrews,and Q.Xu,J. Phys.Chem.A 107,2458(2003).
[47]Q.N.Zhang,W.L.Li,C.Xu,M.H.Chen,M.F.Zhou, J.Li,D.M.Andrada,and G.Frenking,Angew.Chem. Int.Ed.54,11078(2015).
[48]M.F.Zhou,N.Tsumori,Z.H.Li,K.N.Fan.L.Andrews,and Q.Xu,J.Am.Chem.Soc.124,12936 (2002).
[49]M.F.Zhou,Z.X.Wang,P.R.Schleyer,and Q.Xu, Chem.Phys.Chem.4,763(2003).
[50]M.F.Zhou,Q.Xu,Z.X.Wang,and P.R.Schleyer,J. Am.Chem.Soc.124,14854(2002).
[51]H.Braunschweig,R.D.Dewhurst,F.Hupp,M.Nutz, K.Radacki,C.W.Tate,A.Vargas,and Y.Ye,Nature 522,327(2015).
[52]G.J.Wang,C.X.Chi,X.P.Xing,C.J.Ding,and M. F.Zhou,Sci.China Chem.57,172(2014).
[53]G.J.Wang,C.X.Chi,J.M.Cui,X.P.Xing,and M. F.Zhou,J.Phys.Chem.A 116,2484(2012).
[54]A.D.Becke,J.Chem.Phys.98,5648(1993).
[55]C.Lee,W.Yang,and R.G.Parr,Phys.Rev.B 37,785 (1988).
[56]D.E.Woon and T.H.Dunning Jr.,J.Chem.Phys. 100,2975(1994).
[57]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E.Scuseria,M.A.Robb,J.R.Cheeseman,G.Scalmani,V. Barone,B.Mennucci,G.A.Petersson,H.Nakat-suji, M.Caricato,X.Li,H.P.Hratchian,A.F.Iz-maylov, J.Bloino,G.Zheng,J.L.Sonnenberg,M.Hada, M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa,M. Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai, T.Vreven,J.A.Montgomery Jr.,J.E.Peralta,F. Ogliaro,M.Bearpark,J.J.Heyd,E.Brothers,K.N. Kudin,V.N.Staroverov,R.Kobayashi,J.Normand, K.Raghavachari,A.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,N.J.Millam,M. Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo, J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev, A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochter-ski, R.L.Martin,K.Morokuma,V.G.Zakrzewski,G.A. Voth,P.Salvador,J.J.Dannenberg,S.Dapprich,A. D.Daniels,¨O.Farkas,J.B.Foresman,J.V.Ortiz,J. Cioslowski,and D.J.Fox,Gaussian 09,Revision A02, Pittsburgh,PA:Gaussian,Inc.,(2009).
[58]A.D.Becke,Phys.Rev.A 38,3098(1988).
[59]J.P.Perdew,Phys.Rev.B 33,8822(1986).
[60]J.G.Snijders,E.J.Baerends,and P.Vernoojs,At. Data Nucl.Data Tables 26,483(1981).
[61]G.Te Velde,F.M.Bickelhaupt,E.J.Baerends,C. Fonseca Guerra,S.J.A.Van Gisbergen,J.G.Snijders, and T.Ziegler,J.Comput.Chem.22,931(2001).
[62]M.Okumura,L.I.Yeh,J.D.Myers,and Y.T.Lee,J. Chem.Phys.85,2328(1986).
[63]W.H.Robertson and M.A.Johnson,Annu.Rev.Phys. Chem.54,173(2003).
[64]A.M.Ricks,Z.E.Reed,and M.A.Duncan,J.Mol. Spectrosc.266,63(2011).
[65]G.J.Wang,J.M.Cui,C.X.Chi,X.J.Zhou,Z.H.Li, X.P.Xing,and M.F.Zhou,Chem.Sci.3,3272(2012).
[66]J.M.Cui,G.J.Wang,X.J.Zhou,C.X.Chi,Z.H.Li, Z.P.Liu,and M.F.Zhou,Phys.Chem.Chem.Phys 15,10224(2013).
[67]P.Pyykko and M.Atsumi,Chem.Eur.J.15,12770 (2009).
[68]M.P.Mitoraj,A.Michalak,and T.Ziegler,J.Chem. Theory Comput.5,962(2009).
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- ARTICLE E ffi cient Separation of Ar and Kr from Environmental Samples for Trace Radioactive Noble Gas Detection†
- REVIEW Polarization Dependent Time-Resolved Infrared Spectroscopy and Its Applications†
- ARTICLE Reactions of Group V Metal Atoms with Hydrogen Sul fi de:Argon Matrix Infrared Spectra and Theoretical Calculations†
- ARTICLE Structural Dynamics of Phenyl Azide in Light-Absorbing Excited States: Resonance Raman and Quantum Mechanical Calculation Study†
- ARTICLE Structural and Infrared Spectroscopic Study on Solvation of Acetylene by Protonated Water Molecules†
- ARTICLE Excited-State Proton Transfer and Decay in Hydrogen-Bonded Oxazole System:MS-CASPT2//CASSCF Study†