131I难治性分化型甲状腺癌的再分化治疗
2016-04-08程凌霄陈立波
程凌霄,陈立波
上海交通大学附属第六人民医院核医学科,上海 200233
131I难治性分化型甲状腺癌的再分化治疗
程凌霄,陈立波
上海交通大学附属第六人民医院核医学科,上海 200233
[摘要]131I难治性分化型甲状腺癌(radioiodine-refractory differentiated thyroid cancer,RR-DTC)是目前甲状腺癌临床治疗领域的一大难题。维甲酸类药物、过氧化物酶体增殖物激活受体激动剂、DNA甲基化酶抑制剂及组蛋白脱乙酰化酶抑制剂都曾被用于诱导RR-DTC再分化并与131I联合治疗,但疗效并不显著。近年来,随着对RR-DTC分子机制认识的不断深入,靶向治疗等新的再分化治疗策略越来越多地被尝试用于治疗RRDTC。相比之下,分子靶向药物用于诱导RR-DTC重摄碘及介导131I治疗效果较好,可能具有良好的应用前景。
[关键词]甲状腺癌;再分化治疗;靶向药物;131I

陈立波,留德医学博士,上海交通大学附属第六人民医院核医学科主任医师、教授、博士生导师,核医学研究室副主任,上海市抗癌协会甲状腺肿瘤专业委员会委员,中国临床肿瘤学会(CSCO)甲状腺癌专业委员会副主任委员,国家自然科学基金评审专家。
曾任德国海德堡大学与德国癌症研究中心客座科学家、横滨市立大学客座研究员。曾获世界核医学与生物学联盟“杰出贡献奖”、日本核医学会“亚洲青年研究者奖”、上海市“医学科技奖”、上海市“科技启明星”等奖项和荣誉。
主持国家自然科学基金、上海市科技启明星计划等科研课题6项,临床和研究方向为甲状腺疾病的诊治和肿瘤分子影像诊断。以第一作者或通信作者在Thyroid、Journal of Nuclear Medicine、Journal of Clinical Endocrinology and Metabolism等专业期刊发表甲状腺疾病诊治临床和基础研究SCI论文20余篇。
Re-diferentiating therapy of radioiodine-refractory diferentiated thyroid cancer
CHENG Lingxiao,CHEN Libo
(Department of Nuclear Medicine, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai 200233, China)
Correspondence to:CHEN Libo E-mail:libochen888@hotmail.com
[Abstract]Clinical management of radioiodine-refractory diferentiated thyroid cancer(RR-DTC)is extremely di ffi cult.Re-diferentiation compounds, such as retinoids, peroxisome proliferator-activated receptor(PPAR)agonists, DNA methyltransferase inhibitors and histone deacetylase inhibitors, have been used in trials to increase iodine uptake in RR-DTC.However, data on these drugs failed to meet the initial high expectations.In recent years, targeted agents have been increasingly used in pre-clinical and clinical studies to induce re-diferentiation and mediate131I therapy, and the outcomes are encouraging.
[Key words]Thyroid cancer; Re-diferentiating therapy; Targeted agents;131I
分化型甲状腺癌(differentiated thyroid cancer,DTC)主要包括甲状腺乳头状癌(papillary thyroid carcinoma,PTC)和甲状腺滤泡状癌(follicular thyroid carcinoma,FTC)两种病理类型,它们占所有甲状腺癌的80%~90%。DTC的总体预后较好,10年生存率可达85%[1]。多数DTC对131I治疗敏感,手术切除、131I治疗及甲状腺激素抑制的联合疗法(低复发风险组患者不需要行131I治疗)可使此类患者获得长期无病生存。然而,全部DTC患者中2%~5%及有远处转移的DTC中约67%在其自然病程或治疗过程中,肿瘤细胞形态和功能出现退行性变化,失去分化表型[2-3],如促甲状腺激素受体(thyroidstimulating hormone receptor,TSHR)和钠碘同向转运体(natrium iodide symporter,NIS)表达降低及功能异常,使甲状腺素抑制疗法和131I治疗难以奏效。
目前,131I难治性DTC(radioiodine-refractory differentiated thyroid cancer,RR-DTC)的常用定义为:131I显像至少有1个不摄碘病灶或131I治疗后1年内出现病情进展或131I累积剂量超过600 mCi而疾病无缓解[4]。尽管RR-DTC较为少见,但是它具有高侵袭性。对131I治疗敏感的转移性DTC患者10年生存率可达60%,而对转移性RR-DTC患者10年生存率仅为10%[4]。此外,RR-DTC常常伴有多发转移灶,不适于手术和外照射治疗[5];同时,包括细胞毒性药物(如多柔比星的反应率在0%~22%之间,且不良反应较大)在内的全身疗法效果欠佳[6],这使得RRDTC的治疗工作面临很大挑战。
目前认为引起DTC细胞失分化的原因有:① 经131I治疗后,未被杀死的DTC细胞的代谢过程都可能因辐射作用的影响发生改变,特别是Tg的合成和碘代谢易受影响,从而失去摄碘能力;② 在131I治疗前就可能存在具有不同摄碘能力的肿瘤细胞克隆,131I治疗选择性地杀死摄碘能力强的细胞,而摄碘能力差的转移灶DTC细胞的形态和功能均发生明显的改变,细胞摄取131I的功能明显减弱,是影响DTC患者预后的主要原因之一;③ 未经甲状腺全切手术或未经131I去除术后残留甲状腺组织的DTC患者,常会出现局部或远处转移灶肿瘤细胞失分化程度高于原发灶的现象;④ 随着年龄的增加,失分化转移灶的发生率也逐渐增加,65岁以上高达40%。
近年来,再分化治疗(即利用各类药物使肿瘤再分化,提高摄碘率)这一新的治疗策略被尝试用于治疗RR-DTC,取得了一定成效。再分化治疗使131I这一DTC特异性的经典治疗药物被用于治疗RR-DTC成为可能,并可避免长期使用各类靶向药物带来的较大不良反应,且在部分临床前及临床试验中显示出了较好的疗效。现对该领域的研究进展作如下综述。
1 作用于核受体诱导再分化的药物
核受体是一种脂溶性配体依赖转录因子,通过调控靶基因的转录过程,在个体发育中参与多种生理功能的调节,如形态发育、细胞增殖分化、机体稳态维持及高级神经功能控制等,其配体包括激素、胆汁酸、脂质、药物(包括抗癌药)和其他外源性化学物质。核受体的活化开始于细胞质,在细胞质中,大多数未结合配体的核受体与转录辅抑制子及脱乙酰基组蛋白结合,阻止靶基因的转录。核受体与一些药物结合后,其构象发生改变,与辅抑制因子及脱乙酰基组蛋白脱离,并转位入细胞核,与辅活化因子结合成复合物,与自身结合形成同源二聚体,在大多数情况下与视黄醇类X受体(retinoid X receptor,RXR)形成异二聚体。这些异二聚体结合于靶基因的调控区(启动子和增强子)上,启动靶基因转录[7-9]。此类药物包括维甲酸类药物和过氧化物酶体增殖物激活受体(proxisome proliferation-activated receptor,PPAR)激动剂。
1.1 维甲酸类药物
维甲酸类药物是甲状腺癌再分化研究最早用到的药物之一。他们通过作用于维甲酸受体和视黄酸受体等核受体提高甲状腺特异蛋白的表达水平从而提高甲状腺癌细胞的摄碘率;同时降低正常细胞的摄碘率[10]。发展至今,已经有3代维甲酸类药物。第1代药物包括视黄醇、视黄醛和视黄酸(如全反式维甲酸、9-顺视黄酸和13-顺视黄酸)。第2代药物有阿维A酯和阿维A酸。第3代有阿扎罗汀和贝沙罗汀。
以往对甲状腺癌再分化的研究主要集中于13-顺视黄酸和全反式维甲酸等第1代药物上,其临床效果是有限的。在Simon等[11]的研究中,异维甲酸(13-顺视黄酸)可以使40%的RRDTC患者摄碘率提高,而在其他研究[12-13]中,仅少数患者摄碘率得以提高。有关全反式维甲酸的各临床试验结果也很不一致[14-15]。在最近的全反式维甲酸再分化试验的报道中,55%研究对象有摄碘率的提高,但此次研究规模很小[15]。值得注意的是,在Simon等[11]的研究中,一些患者是否存在摄碘率提高与131I治疗是否有效并不一致。所以,该疗效能否部分归因于维甲酸介导再分化后131I的治疗作用尚不明确,今后的临床研究还需要增加对照组。另外2种第1代药物(9-顺视黄酸及视黄醇)的研究目前还仅局限于体外,后者被证实可以提高细胞系的摄碘率[16]。
1.2 PPAR激动剂
PPAR是一类由配体激活的核转录因子,属于维甲酸-类固醇-甲状腺素核因子受体超家族。PPAR家族包括PPAR-α、PPAR-δ和PPAR-γ3类受体,其中PPAR-γ分布广泛,对维持甲状腺细胞正常生长、增殖及分化具有较为重要的作用。PPAR-γ在与配体结合后,和RXR形成异源二聚体,继而结合靶基因启动子区过氧化物酶体增殖物反应元件(PPAR-γ response element,PPRE),激活目的基因转录而对体内多种生理及病理过程发挥调控作用[17]。
PAX8-PPAR-γ染色体重排发生于36%~45% 的FTC和37.5%的滤泡型甲状腺乳头状癌(follicular variant of papillary thyroid cancer,FVPTC)[18-19],它是由于t(2;3)(q13;p25)染色体平衡易位,导致编码转录因子PAX8的基因与PPAR-γ基因A到F区域的融合。PAX8-PPAR-γ重排导致PAX8-PPAR-γ融合蛋白(PAX8-PPAR-γ fusion protein,PPFP)表达增高。PPFP能强有力地抑制PPAR-γ与PPRE结合,从而扰乱甲状腺细胞的正常生长与分化,促使细胞快速增殖与异常分化[20]。PPAR激动剂(如罗格列酮、吡格列酮和曲格列酮等)与PPAR-γ结合,后者与RXR形成异质二聚体,继而结合靶基因启动子区PPRE,激活目的基因转录,进一步促进靶基因PTEN的表达,而使甲状腺癌细胞中异常激活的PI3K通路受到抑制[21]。
罗格列酮诱导甲状腺癌再摄碘的报道始于10年前[22],Elola等[21]和 Elias等[24]分别报道过罗格列酮使不摄碘甲状腺癌转移灶重新摄碘的患者。在另外2次临床试验中[25-26],经罗格列酮治疗6周以后,分别有40%和26%的患者摄碘率有所提高。但在以上所有研究中,并没有发现肿瘤负荷的减小。其他PPAR激动剂(如曲格列酮)只在体外试验中被证实可以提高甲状腺癌细胞的摄碘率[27],且曲格列酮的效果优于吡格列酮和环格列酮[28]。值得一提的是,Tepmongkol等[26]的免疫组织化学分析显示,甲状腺癌细胞中PPAR-γ受体高表达的患者会在罗格列酮预处理后出现重摄碘现象,而低表达或不表达PPAR-γ受体的患者极少会重新摄碘。
2 针对表观遗传学改变的药物
表观遗传学改变,即诸如DNA甲基化或组蛋白去乙酰化之类的遗传物质修饰,在肿瘤细胞中这些改变会导致细胞分化相关基因的沉默。所以,DNA甲基化酶抑制剂(DNA methyltransferanse inhibitor,DMI)和组蛋白去乙酰化酶抑制剂(histone deacetylase inhibitor,HDACI)可以使甲状腺癌细胞中的这些基因再表达,从而诱导细胞再分化并提高细胞摄碘率。
2.1 DNA甲基化酶抑制剂
DNA甲基化是指在DNA甲酰转移酶作用下,以S-腺苷甲硫氨酸为甲基供体,将甲基转移到胞嘧啶鸟嘌呤二核苷酸的胞嘧啶的第5位碳原子上的过程。CpG岛为富含胞嘧啶鸟嘌呤二核苷酸的区域,位于基因的5’端区域,大多数CpG岛是低甲基化的。而肿瘤细胞普遍存在甲基化失衡的状况。转录起点附近基因启动子甲基化时,常会造成基因沉默,使得一些重要的基因丧失功能。在甲状腺癌细胞中,启动子区域的高度甲基化使NIS表达减少,细胞摄碘率因此降低;此外,甲状腺转录因子-1(thyroid transcription factor-1,TTF-1)是Tg、TPO、TSHR、PDS和NIS等甲状腺特异基因的重要转录因子,TTF-1基因的甲基化使其表达减少,TTF-1与启动子的结合减少,最终导致上述多个基因的沉默[29]。
Venkataraman等[29]的研究发现,DMI药物5-azacytidine虽然能使KAT-1、KAT-5、KAT-10 及NPA-87这4个甲状腺乳头状癌细胞株的hNIS 的mRNA再表达,但在MRO87和WRO82(FTC)及KAT-7(PTC)中却未得到相同结果。在5-azacytidine等药物对细胞株Nthy-ori 3-1及B-CPAP(PTC)的Tg、TPO、TSHR、NIS的mRNA表达及摄碘率影响的研究中,正常甲状腺细胞和甲状腺肿瘤细胞的NIS基因的表达在药物处理后均未见提高,肿瘤细胞的表达水平甚至出现降低,而摄碘率提高仅见于正常细胞[30]。上述试验中的阴性结果可能是因为存在转录后修饰的一系列复杂机制。另一种DMI(5-aza-20-deoxycytidine)也已在体外被证实可以提高PTC、FTC和未分化型甲状腺癌细胞中TTF-1的表达水平[31]。
2.2 组蛋白脱乙酰化酶抑制剂
组蛋白是染色质中核小体(真核细胞中染色质的基本结构)的主要结构元件。组蛋白被修饰所引起的染色质结构改变在真核生物基因表达调控中发挥着重要作用,这些修饰包括乙酰化、甲基化、磷酸化和泛素化等。其中,组蛋白乙酰化尤为重要。核小体中的组蛋白氨基末端富含赖氨酸,通过与乙酰基结合或解构来改变DNA的构象[32]。组蛋白的乙酰化状态由2种酶的作用决定:组蛋白乙酰化转移酶(histone acetyltransferase,HAT)和HDAC。其中,HDAC使得DNA与核心组蛋白紧密结合,阻碍基本转录单位蛋白质复合物进人启动子结合位点,抑制转录功能。研究发现,HDAC功能紊乱可导致组蛋白乙酰化的不平衡,染色质结构改变以及细胞生长、分化和凋亡相关基因表达受抑制[33-34]。而HDACI的抗肿瘤机制就表现在重新平衡癌细胞中紊乱的组蛋白乙酰化状态,阻滞癌细胞生长、诱导凋亡和促进分化。
非特异性HDACI有伏立诺他、丙戊酸、帕比司他和曲古抑菌素A;特异性HDACI包括罗咪酯肽、apicidin、恩替诺特和APHA。这些药物在体外试验中多可提高NIS的表达水平或提高细胞摄碘率[16],但在临床试验中并未见较好疗效。对伏立诺他诱导甲状腺癌再分化的临床研究开展较早,在2005年的Ⅰ期临床试验中,在5位受试者中有1位摄碘率有所提高[35]。2013年,罗咪酯肽诱导再分化的Ⅱ期临床试验中,在20例患者中仅有2例摄碘率提高,但这2例患者在后续131I治疗中未见客观治疗反应[36]。帕比司他是一种新型HDACI,在2015年2月被美国食品和药品管理局(Food and Drug Administration,FDA)批准用于多发性骨髓瘤的治疗[37]。在2013年的体外研究中,帕比司他被证实可以提高NIS的表达水平并提高131I的细胞毒性作用[38],相应的Ⅱ期临床试验(NCT01013597)正在进行[39]。
3 分子靶向药物
RET-RAS-RAF-MEK-MAPK/ERK与PI3KAKT-mTOR这2条信号通路传递甲状腺细胞生长、增殖和分化信号,它们的异常在甲状腺癌的发生、发展中有着重要地位。在DTC中一些常见遗传学改变,如BRAF和RAS突变,RET/PTC重排,导致信号转导通路的组成性激活,最终造成钠碘同向转运体的低表达及质膜定位量的减少[40-41]。这些异常激活通路与NIS功能减低的关系预示着抑制这些通路或许是重获NIS功能的有效方法,直接作用于信号转导通路诱导再分化的策略应运而生。
临床前和临床研究结果都已显示出分子靶向药物提高甲状腺癌细胞摄碘率的作用(表1)。体外研究[42-43]表明,作用于MAPK和PI3KAKT通路上的BRAF、MEK和AKT等靶点可以提高甲状腺基因的表达水平和细胞的摄碘率,且这种效应在TSH的介导下可以得到增强。随后的一项体内试验证实了这种作用[44]。在这项动物实验中,转基因小鼠在药物诱导下出现BRAFV600E突变并发生甲状腺癌,在分别给与MEK抑制剂PD0325901和BRAF抑制剂PLX4720后,肿瘤重获对放射碘治疗的敏感性[44]。与之类似,在我们的体外研究中[45],分别用索拉非尼和卡博替尼处理带有RET/PTC重排的BHP 2-7细胞系后,碘代谢相关基因表达增加而1型和3型葡萄糖转运体的表达受到抑制,同时发现细胞的碘摄取提高而糖代谢水平降低,相应的体内试验正在进行。
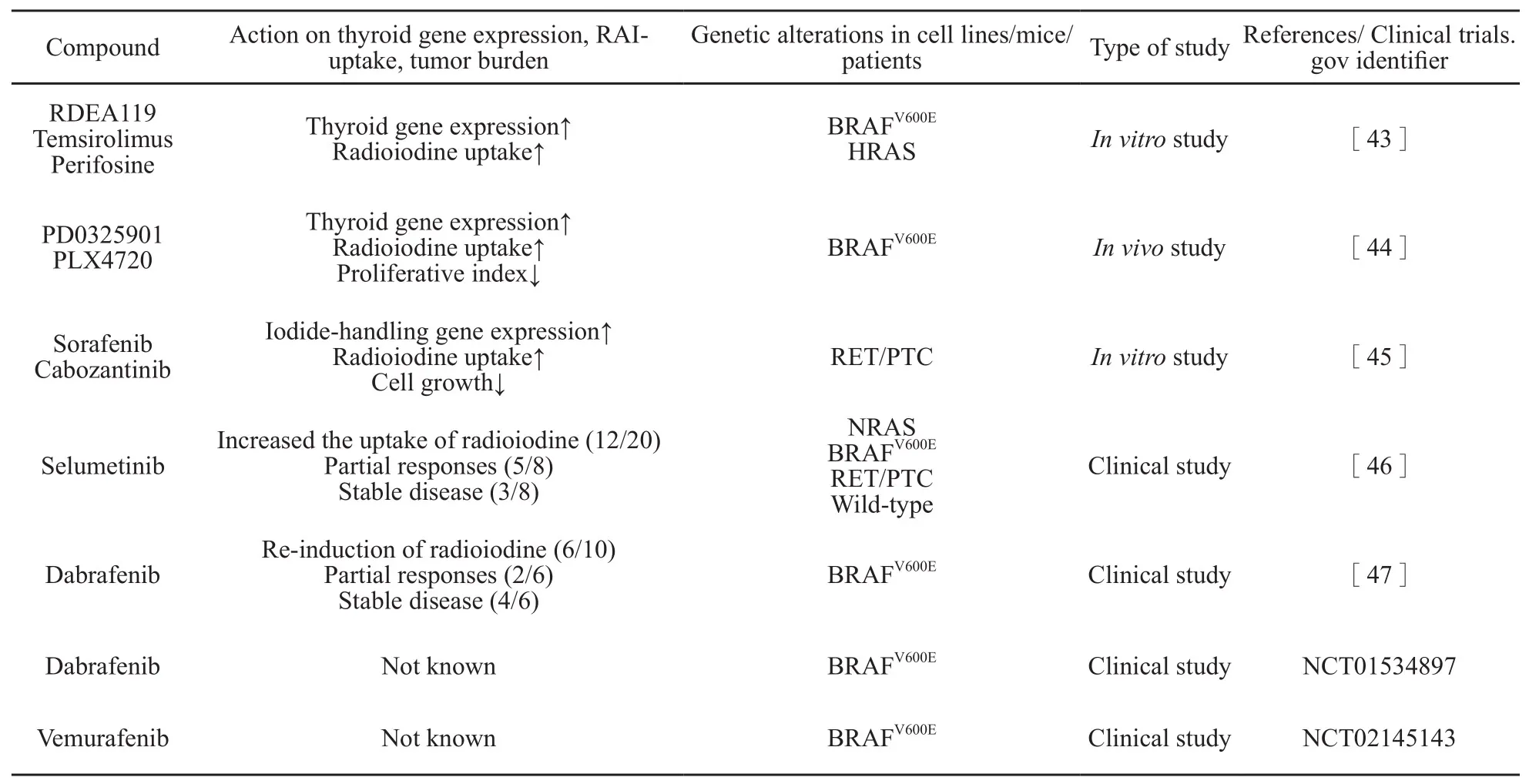
表 1 用于分化型甲状腺癌再分化治疗的靶向药物Tab.1 Current state of targeted drugs for re-diferentiating therapy of diferentiated thyroid carcinoma
近期一项用司美替尼和131I联合治疗甲状腺癌的临床试验[46]获得了喜人的结果。在20例可评估RR-DTC患者中有12例患者的碘摄取有所提高,其中8例患者124I PET/CT扫描结果示摄碘大于20 Gy,他们接受了131I治疗且都有治疗反应的影像学证据。在这20例患者中,携带NRAS突变的所有个体(5例)肿瘤摄碘率都有所提高且其中4例达到部分缓解而在携带BRAF突变的个体中却效果欠佳,这引发了对信号通路调节及个体化治疗的进一步思考。正在进行的司美替尼联合131I治疗甲状腺癌术后高危患者的Ⅲ期临床试验(NCT01843062)将会提供更多信息。在用达拉非尼(BRAF抑制剂)诱导再分化的临床试验中,达拉非尼使60%(6/10)有远处转移并带有BRAF突变的PTC患者重新摄碘,随后,这6例患者全部接受了131I(150 mCi)治疗,其中2例患者获得部分反应,4例患者3个月随访时病情稳定,共有4例患者Tg水平下降[47]。目前,还有2项分别应用达拉非尼和维罗非尼诱导带有BRAF突变的PTC病灶再分化的临床试验正在进行(NCT01534897 和NCT02145143)。
鉴于131I是一种相对安全的治疗手段,在碘难治的患者中,将分子靶向药物与131I联合应用可以避免长期单独使用分子靶向药物带来的较大不良反应,发挥131I这一DTC经典治疗方法安全、快捷的优势。另外,可喜的是,在骨和淋巴结转移灶等对靶向治疗反应欠佳的部位也可以出现摄碘率的提高[46]。综上所述,尽管有关分子靶向药物诱导RR-DTC再分化并提高131I治疗的反应率的证据有限,但是分子靶向药物和131I联合目前看来是一种有前途的治疗策略。随着越来越多针对不同靶点的新药不断进入临床试验验证,其疗效值得期待。
为获知这些药物诱导再摄碘的程度,需要开展更大规模的临床研究。就目前2项碘难治患者再分化的研究而言,入组的标准是有所区别的,在今后的研究中统一入组标准可使研究结果有更明确的意义。在一些前期研究中,摄碘率的测量采用传统131I扫描[47],而能够精确定量的124I PET/CT或许可以在以后的研究中更多应用以更好评估疗效,并有助于为后续的131I用药制定剂量。对于DTC这种生长相对缓慢的肿瘤,这种再分化治疗能否延长生存期也需要更长时间的随访。此外,疗效归因于单纯分子靶向药物的抗肿瘤作用还是因它们介导再分化后131I的作用也还有待探讨。值得注意的是,上述再分化治疗的临床试验中分子靶向药物治疗时间均较短,疗效也是在撤药后出现,高度提示疾病控制得益于131I的作用。
针对特定突变的个体化疗法也值得探索。临床研究中只有部分BRAF突变的患者能够被诱导重摄碘,其中机制尚待深入研究。已知BRAF突变介导的摄碘率降低只是部分依赖于ERK水平的改变,用BRAF或MEK抑制剂来抑制异常激活的MAPK通路并不能完全使细胞恢复失分化前的摄碘水平,而且RAF和MEK抑制剂在通路下游的抑制作用会因为反馈机制而减弱[44,48-49]。今后,MAPK通路抑制剂和其他激酶抑制剂联合应用来对抗反馈作用以取得更强的再分化效果值得期待。
4 小结
多类靶向药物可以诱导RR-DTC的再分化。维甲酸类药物、过氧化物酶体增殖物激活受体激动剂、DNA甲基化酶抑制剂及组蛋白脱乙酰化酶抑制剂等药物在体外试验中可以使RR-DTC细胞再分化,不同程度地提高NIS和TSHR的表达水平并提高摄碘率,但它们的临床试验结果总体不乐观。分子靶向药物诱导RR-DTC再分化并提高131I治疗反应率的临床研究尚不充分,但近期的试验结果已让RR-DTC的治疗见到新的曙光。
[参考文献]
[1]EUSTATIA-RUTTEN C F, CORSSMIT E P, BIERMASZ N R, et al.Survival and death causes in differentiated thyroid carcinoma[J].J Clin Endocrinol Metab, 2006, 91(1):313-319.
[2]RIVERA M, GHOSSEIN R A, SCHODER H, et al.Histopatholologic characterization of radioactive iodinerefractory fluorodeoxyglucose-positron emission tomographypositive thyroid carcinoma[J].Cancer, 2008, 113(1):48-56.
[3]BROSE M S, NUTTING C M, JARZAB B, et al.Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer:a randomised, double-blind, phase 3 trial[J].Lancet, 2014, 384(9940):319-328.
[4]DURANTE C, HADDY N, BAUDIN E, et al.Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma:benefits and limits of radioiodine therapy[J].J Clin Endocrinol Metab, 2006, 91(8):2892-2899.
[5]SAMPSON E, BRIEDEY J D, LE L W, et al.Clinical management and outcome of papillary and follicular(differentiated)thyroid cancer presenting with distant metastasis at diagnosis[J].Cancer, 2007, 110(7):1451-1456.
[6]SHERMAN S I.Cytotoxic chemotherapy for differentiated thyroid carcinoma[J].Clin Oncol, 2010, 22(6):464-468.
[7]SYNOLD T W, DUSSAULT I, FORMAN B M.The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux[J].Nat Med, 2001, 7(5):584-590.
[8]JONES S A, MOORE L B, SHENK J L, et al.The pregnane X receptor:a promiscuous xenobiotic receptor that has diverged during evolution[J].Mol Endocrinol, 2000, 14(1):27-39.
[9]TAKESHITA A, TAGUCHI M, KOIBUCHI N, et al.Putative role of the orphan nuclear receptor SXR(steroid and xenobiotic receptor)in the mechanism of CYP3A4 inhibition by xenobiotics[J].J Biol Chem, 2002, 277(36):32453-32458.
[10]JEONG H, KIM Y R, KIM K N, et al.Effect of all-transretinoic acid on sodium/iodide symporter expression, radioiodine uptake and gene expression profiles in a human anaplastic thyroid carcinoma cell line[J].Nucl Med Biol, 2006, 33(7):875-882.
[11]SIMON D, KÖRBER C, KRAUSCH M, et al.Clinical impact of retinoids in redifferentiation therapy of advanced thyroid cancer:final results of a pilot study[J].Eur J Nucl Med Mol Imaging, 2002, 29(6):775-782.
[12]KIM W G, KIM E Y, KIM T Y, et al.Redifferentiation therapy with 13-cis retinoic acids in radioiodine-resistant thyroid cancer[J].Endocr J, 2009, 56(1):105-112.
[13]HANDKIEWICZ-JUNAK D, ROSKOSZ J, HASSE-LAZAR K, et al.13-cis-retinoic acid re-differentiation therapy and recombinant human thyrotropin-aided radioiodine treatment of non-functional metastatic thyroid cancer:a single-center, 53-patient phase 2 study[J].Thyroid Res, 2009, 2(1):8.
[14]ZHANG Y, JIA S, LIU Y, et al.A clinical study of all-transretinoid-induced differentiation therapy of advanced thyroid cancer[J].Nucl Med Commun, 2007, 28(4):251-255.
[15]DAMLE N, PATNECHA M, KUMAR P, et al.Retinoic acid therapy in patients with radioiodine negative differentiated thyroid cancer and clinical or biochemical evidence of disease:An initial experience[J].Indian J Nucl Med, 2011, 26(3):144-148.
[16]FRÖHLICH E, BROSSART P, WAHL R.Induction of iodide uptake in transformed thyrocytes:a compound screening in cell lines[J].Eur J Nucl Med Mol Imaging, 2009, 36(5):780-790.
[17]NIKIFOROVA M N, BIDDINGER P W, CAUDILL C M, et al.PAX8-PPARgamma rearrangement in thyroid tumors:RTPCR and immunohistochemical analyses[J].Am J Surg Pathol, 2002, 26(8):1016-1023.
[18]CASTRO P, REBOCHO A P, SOARES R J, et al.PAX8-PPARgamma rearrangement is frequently detected in the follicular variant of papillary thyroid carcinoma[J].J Clin Endocrinol Metab, 2006, 91(1):213-220.
[19]NIKIFOROVA M N, LYNCH R A, BIDDINGER P W, et al.RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors:evidence for distinct molecular pathways in thyroid follicular carcinoma[J].J Clin Endocrinol Metab, 2003, 88(5):2318-2326.
[20]KROLL TG, SARRAF P, PECCIARINI L, et al.PAX8-PPAR gamma fusion oncogene in human thyroid carcinoma[J].Science, 2000, 289(5483):1357-1360.
[21]KAPITEIJN E, SCHNEIDER T C, MORREAU H, et al.New treatment modalities in advanced thyroid cancer[J].Ann Oncol, 2012, 23(1):10-18.
[22]PHILIPS J C, PETITE C, WILLI J P, et al.Effect of peroxisome proliferator-activated receptor gamma agonist, rosiglitazone, on dedifferentiated thyroid cancers.[J].Nucl Med Commun, 2004, 25(12):1183-1186.
[23]ELOLA M, YOLDI A, EMPARANZA J I, et al.Redifferentiation therapy with rosiglitazone in a case of differentiated thyroid cancer with pulmonary metastases and absence of radioiodine uptake[J].Rev Esp Med Nucl, 2011, 30(4):241-243.
[24]ELIAS A N, LIZOTTE P.Enhanced radioiodine uptake in a patient with poorly differentiated papillary thyroid cancer after treatment with rosiglitazone[J].Clin Nucl Med, 2006, 31(9):517-519.
[25]KEBEBEW E, PENG M, REIFF E, et al.A phase Ⅱ trial of rosiglitazone in patients with thyroglobulin-positive and radioiodine-negative differentiated thyroid cancer[J].Surgery, 2006, 140(6):960-966.
[26]TEPMONGKOL S, KEELAWAT S, HONSAWEK S, et al.Rosiglitazone effect on radioiodine uptake in thyroid carcinoma patients with high thyroglobulin but negative total body scan:a correlation with the expression of peroxisome proliferatoractivated receptor-gamma[J].Thyroid, 2008, 18(7):697-704.
[27]FRÖHLICH E, MACCHICAO F, WAHL R.Action of thiazolidinediones on differentiation, prolifration and apoptosis of normal and transformed thyrocytes in culture[J].Endocr Relat Cancer, 2005, 12(4):1-13.
[28]MARTELLI M, IULIANO R, LE PERA I, et al.Inhibitory effects of peroxisome poliferator-activated receptor gamma on thyroid carcinoma cell growth[J].J Clin Endocrinol Metab, 2002, 87(10):4728-4735.
[29]VENKATARAMAN G M, YATIN M, MARCINEK R, et al.Restoration of iodide uptake in dedifferentiated thyroid carcinoma:relationship to human Na+/I-symporter gene methylation status[J].J Clin Endocrinol Metab, 1999, 84(7):2449-2457.
[30]TUNCEL M, AYDIN D, YAMAN E, et al.The comparative effects of gene modulators on thyroid-specific genes and radioiodine uptake[J].Cancer Biother Radiopharm, 2007, 22(3):443-449.
[31]KONDO T, NAKAZAWA T, MA D, et al.Epigenetic silencing of TTF-1/NKX2-1 through DNA hypermethylation and histone H3 modulation in thyroid carcinomas[J].Lab Invest, 2009, 89(7):791-799.
[32]LUGER K.Structure and dynamic behavior of nucleosomes[J].Curr Opin Genet Dev, 2003, 13(2):127-135.
[33]DUNCAN E M, MURATORE-SCHROEDER T L, COOK R G, et al.Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation[J].Cell, 2008, 135(2):284-294.
[34]GLASER K B.HDAC inhibitors:clinical update and mechanism-based potential[J].Biochem Pharmacol, 2007, 74(5):659-671.
[35]KELLY W K, O’CONNOR O A, KRUG L M, et al.Phase Ⅰstudy of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer[J].J Clin Oncol, 2005, 23(17):3923-3931.
[36]SHERMAN E J, SU Y B, LYALL A, et al.Evaluation of romidepsin for clinical activity and radioactive iodine reuptake in radioactive iodine-refractory thyroid carcinoma[J].Thyroid, 2013, 23(5):593-599.
[37]No authors listed.Panobinostat approved for multiple myeloma[J].Cancer Discov, 2015, 5(5):OF4.
[38]PUGLIESE M, FORTUNATI N, GERMANO A, et al.Histone deacetylase inhibition affects sodium iodide symporter expression and induces(131)Ⅰ cytotoxicity in anaplastic thyroid cancer cells[J].Thyroid, 2013, 23(7):838-846.
[39]ANDERSON R T, LINNEHAN J E, TONGBRAM V, et al.Clinical, safety, and economic evidence in radioactive iodinerefractory differentiated thyroid cancer:a systematic literature review[J].Thyroid, 2013, 23(4):392-407.
[40]LIU M, RUAN M, CHEN L.Update on the molecular diagnosis and targeted therapy of thyroid cancer[J].Med Oncol, 2014, 31(6):973-973.
[41]DOHAN O, BALOCH Z, BANREVI Z, et al.Rapid communication:predominant intracellular overexpression of the Na(+)/I(-)symporter(NIS)in a large sampling of thyroid cancer cases[J].J Clin Endocrinol Metab, 2001, 86(6):2697-2700.
[42]LIU D, HU S, HOU P, et al.Suppression of BRAF/MEK/MAP kinase pathway restores expression of iodide-metabolizing genes in thyroid cells expressing the V600E BRAF mutant[J].Clin Cancer Res, 2007, 13(4):1341-1349.
[43]HOU P, BOJDANI E, XING M.Induction of thyroid gene expression and radioiodine uptake in thyroid cancer cells by targeting major signaling pathways[J].J Clin Endocrinol Metab, 2010, 95(2):820-828.
[44]CHAKRAVARTY D, SANTOS E, RYDER M, et al.Smallmolecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation[J].J Clin Invest, 2011, 121(12):4700-4711.
[45]RUAN M, LIU M, DONG Q, et al.Iodide- and glucosehandling gene expression regulated by sorafenib or cabozantinib in papillary thyroid cancer[J].J Clin Endocrinol Metab, 2015, 100(5):1771-1779.
[46]HO A L, GREWAL R K, LEBOEUF R, et al.Selumetinibenhanced radioiodine uptake in advanced thyroid cancer[J].N Engl J Med, 2013, 368(7):623-632.
[47]ROTHENBERG S, MCFADDEN D G, PALMER E L, et al.Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib[J].Clin Cancer Res, 2015, 21(5):1028-1035.
[48]RIESCO-EIZAGUIRRE G, RODRIGUEZ I, DE LA VIEJA A, et al.The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer[J].Cancer Res, 2009, 69(21):8317-8325.
[49]MONTERO-CONDE C, RUIZ-LLORENTE S, DOMINGUEZ J M, et al.Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas[J].Cancer Discov, 2013, 3(5):520-533.
收稿日期:(2015-11-13 修回日期:2015-12-28)
通信作者:陈立波 E-mail:libochen888@hotmail.com
基金项目:国家自然科学基金(81271609);上海市科技启明星(12QH1401600)。
中图分类号:R736.1
文献标志码:A
文章编号:1007-3639(2016)01-0035-08
DOI:10.3969/j.issn.1007-3969.2016.01.006
