UPLC/Q-TOF-MS 方法测定黄芪皂苷Ⅰ、Ⅱ、Ⅳ含量并研究水解对黄芪皂苷类成分的影响
2016-04-06韩旭阳曾祖平首都医科大学附属北京中医医院北京市中医研究所北京100010
韩旭阳 曾祖平 彭 冰 王 宏 张 苍(首都医科大学附属北京中医医院,北京市中医研究所,北京,100010)
UPLC/Q-TOF-MS 方法测定黄芪皂苷Ⅰ、Ⅱ、Ⅳ含量并研究水解对黄芪皂苷类成分的影响
韩旭阳 曾祖平 彭 冰 王 宏 张 苍
(首都医科大学附属北京中医医院,北京市中医研究所,北京,100010)
摘要本实验采用UPLC/Q-TOF-MS方法结合多变量统计分析(PCA、OPLS-DA),分析不同批次黄芪饮片碱洗前后黄芪皂苷类成分变化趋势及原因。并对黄芪皂苷Ⅰ、Ⅱ、Ⅳ进行含量测定。为以黄芪皂苷类成分作为指标成分进行黄芪饮片及含黄芪中成药质量研究提供了实验依据。本实验采用ACQUITY UPLC C18色谱柱以乙腈-甲酸(1 mL/L)水溶液作为流动相进行梯度洗脱,流速0. 45 mL/min,柱温40℃。使用ESI电离源在正、负离子模式下采集数据,应用Masslynx 4. 1质谱工作站软件进行分多变量统计分析(PCA、OPLS-DA)。明确了黄芪饮片供试品制备过程氨水洗涤步骤能够使黄芪皂苷Ⅰ、Ⅱ水解脱乙酰基转化为黄芪皂苷Ⅳ。同时应用UPLC/Q-TOF-MS方法测定了黄芪饮片中黄芪皂苷Ⅰ、Ⅱ、Ⅳ含量,该方法简单、快速、重现性较好,可用于黄芪饮片中该三个成分的含量测定。本实验为以黄芪皂苷类成分作为指标成分进行黄芪饮片及含黄芪中成药质量研究提供了实验依据。
关键词黄芪;碱洗;UPLC/Q-TOF-MS黄芪皂苷Ⅰ、Ⅱ、Ⅳ
UPLC/Q-TOF-MS-Based Chemical Profiling Approach in Studying the Effects of Amic Solution Hydrolysis on Extraction of Radix Astragali
Han Xuyang,Peng Bing,Wang Hong,Zhang Cang,Zeng Zuping
(Beijing Institute of Traditional Chinese Medicine,Beijing Hospital of Traditional Chinese Medicine Affiliated to Capital Medical University,Beijing 100010,China)
Abstract In this study,UPLC/Q-TOF-MS combined with multivariate statistical analysis(PCA,OPLS-DA)were used to investigate the effects of amic solution hydrolysis to extract of Radix astragalus . The analyses revealed that astragalosideⅣis formed during sample preparation from acylated astragalosides like astragaloside I and astragaloside II when using amic solution to deal with the extract. Simultaneously,UPLC/Q-TOF-MS were used to determine the content of astragalosideⅠ,Ⅱ,Ⅳof Radix astragali. This method could evaluate the quality of Radix Astragali effectively,and it is simple,sensitive and reliable.
Key Words Radix astragali;Amic solution hydrolysis;UPLC/Q-TOF-MS astragalosideⅠ,Ⅱ,Ⅳ
黄芪,又称黄耆,李时珍曰:“耆,长也。黄耆色黄,为补药之长,故名。”[1]其药性甘温,入脾肺经。功能补气升阳,益卫固表,利水消肿,托疮生肌,享有“补气之长”“疮家圣药”以及“治疗气虚水肿尿少之要药”等美称。既善补脾肺之气,升举阳气,用于脾肺气虚诸证。又能托毒生肌,善治气虚津亏之疮痈脓成不溃等。现代研究表明黄芪煎剂和黄芪多糖能促进DNA、RNA和蛋白质合成,提高血浆和组织中cAMP和cGMP含量、增强免疫功能。有保肝、改善肾功能、利尿、改善血流变性,促进造血功能、抗衰老、抗病毒、抗肿瘤等作用[2]。黄芪的主要活性成分为皂苷及黄酮类等。中国药典收录黄芪为豆科多年生草本蒙古黄芪Astragalus menbranaceus(Fisch)Bge. var. mongholicus(Bge.)Hsiao或膜荚黄芪Astragalus menbranaceus(Fisch)Bge.的干燥根[3]。并应用HPLC-ELSD法对黄芪药材及饮片的黄芪甲苷、毛蕊花异黄酮葡萄糖苷进行了含量测定。但由于黄芪甲苷含量较低,多数文献在测定黄芪中黄芪甲苷含量时供试品处理过程较为复杂。
超高效液相色谱是近年发展起来的分离技术,具有超高的分离度、超高的灵敏度、超高压力等特点,在中药甚至复方等复杂体系的分析与分离上具有明显的优势。四级杆-飞行时间串联质谱(Q-TOFMS)是高分辨质谱,能够对Marker进行定性与定量分析,其显著特点是高选择性、高灵敏度,不仅能得到较高质量的一级至多级质谱图且精确化合物质核比到小数点后四位。因此,超高效液相色谱与飞行时间质谱联用技术(UPLC/Q-TOF-MS)已成为目前中药复杂体系中活性成分的快速分离和鉴定的有力手段[4-5]。
本实验应用超高效液相色谱-四级杆串联飞行时间质谱联用技术,结合多变量统计分析如主成分分析(PCA)、正交偏最小二乘法(OPLS-DA)探讨黄芪饮片供试品溶液碱洗前后黄芪皂苷类成分含量的变化趋势及原因。同时对黄芪饮片中黄芪皂苷Ⅰ、Ⅱ、Ⅳ进行含量测定,该方法简单,重现性好。本实验为以黄芪皂苷类成分作为指标成分对黄芪饮片进行质量控制研究提供了实验依据。
1 资料与方法
1. 1 一般资料 ACQUITY UIPLC系统(Waters公司);Xevo G2 Q-TOF质谱仪(Waters公司),装有Look-spray接口;离子源电喷雾(ESI);Masslynx 4. 1软件(Waters公司);XS205专业型分析天平-超越系列(Mettler Toledo);MilliQ水纯化系统(Millipore)。
1. 2 实验试剂 甲醇、乙腈均为LC-MS级,购于美国Themo Fisher公司;甲酸购于美国Sigma公司;水为自制超纯水。
1. 3 受试药物 黄芪饮片均由实验室购自北京、山西、天津、江苏等地,经北京市中医研究所中药室何薇主任药师鉴定均豆科多年生草本蒙古黄芪Astragalus menbranaceus(Fisch)Bge. var. mongholicus (Bge.)Hsiao或膜荚黄芪Astragalus menbranaceus (Fisch)Bge.的干燥根。对照品黄芪皂苷Ⅳ(黄芪甲苷)(110781-200613)购自中国食品药品检定研究院。黄芪皂苷Ⅰ、Ⅱ购自上海同田生物科技有限公司(利用NMR等波谱技术鉴定了化学结构,经HPLC面积归一化法检测,纯度均大于98%)。
2 方法
2. 1 供试品溶液的制备 黄芪饮片粉碎成粉过50目筛,备用。
2. 2 制备未碱洗供试品溶液 称取黄芪粉末1. 000 g于250 mL具塞三角瓶中,加100 mL甲醇,50℃超声提取40 min,静置至室温,过滤,残渣加100 mL甲醇,再次50℃超声提取40 min,静置至室温,过滤,合并2次滤液,定容至200 mL容量瓶中。0. 22 μm微孔滤膜过滤,作为未碱洗供试品备用。
2. 3 制备碱洗供试品溶液 取以上未碱洗供试品溶液100 mL,水浴蒸干,残渣加25 mL水溶解,水饱和正丁醇振摇提取4次,25 mL/d,合并四次正丁醇萃取液,氨水洗涤2次,40 mL/次,静置分层,氨水层弃去,洗涤后的正丁醇层水浴蒸干,残渣加100 mL甲醇溶解定容0. 22 μm微孔滤膜过滤,作为碱洗供试品备用。
2. 4 制备对照品溶液 精密称取黄芪皂苷Ⅰ对照品,加甲醇定容为浓度0. 022 6 mg/mL的溶液,作为及黄芪皂苷Ⅰ对照品溶液A备用,吸取一定量黄芪皂苷Ⅰ对照品溶液A,稀释10倍制成浓度为0. 002 26 mg/mL的溶液,作为黄芪皂苷Ⅰ对照品溶液B备用;精密称取黄芪皂苷Ⅱ对照品,加甲醇制成浓度为0. 002 30 mg/mL的溶液,作为及黄芪皂苷Ⅱ对照品溶液A备用,吸取一定量黄芪皂苷Ⅱ对照品溶液A,稀释2倍制成浓度为0. 001 15 mg/mL的溶液,作为黄芪皂苷Ⅱ对照品溶液B备用;精密称取黄芪皂苷Ⅳ对照品,加甲醇制成浓度为0. 0098 mg/mL的溶液,作为及黄芪皂苷Ⅳ对照品溶液A备用,吸取一定量黄芪皂苷Ⅳ对照品溶液A,稀释10倍制成浓度为0. 000 98 mg/mL的溶液,作为黄芪皂苷Ⅳ对照品溶液B备用,吸取一定量黄芪皂苷Ⅳ对照品溶液1,稀释50倍制成浓度为0. 000 196 mg/mL的溶液,作为黄芪皂苷Ⅳ对照品溶液C备用。
2. 5 色谱分析条件 ACQUITY UPLC BEH C18色谱柱(2. 1 mm×100 mm,1. 7 μm);柱温40℃;流速0. 45 mL/min;进样量3 L;流动相:(A)为乙腈,(B)为1 mL/L甲酸溶液,按以下条件进行梯度洗脱:0-5 min:0~10%A,5~7. 5 min:10%A~100%A,7. 5~10 min:100%A~10%A,10~13 min:10%A。
2. 6 质谱条件 ESI,分别在ESI+、ESI-模式下扫描,Sampling Cone:40 V,Capilary:3. 0 kV和2. 5 kV,Desolvation temperatures:350℃,Source temperatures:100℃,Desolvation Gas Flow 800 L/h,Cone Gas Flow:50 L/h,质量扫描范围:m/z 50~1500,数据采集速率:0. 2 s/scan。采用质量锁定(Lock-mass)技术测定准确质量数,2 ng/L的亮氨酸-脑啡肽(LE,ESI+:m/z 556. 2771,ESI-:m/z554. 2615)溶液为质量锁定溶液,其流速为10 μL/min,切换频率为20 s/次。
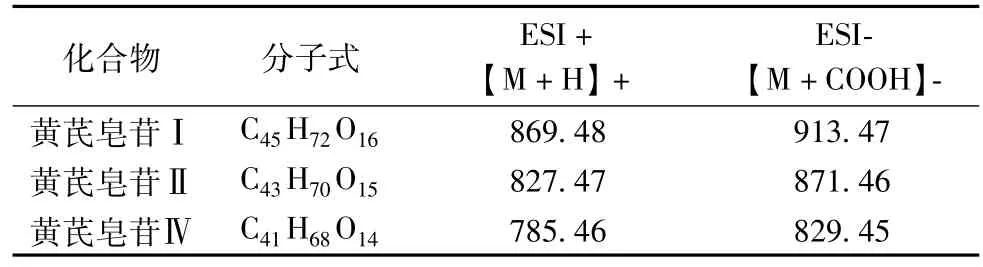
表1黄芪皂苷Ⅰ、Ⅱ、Ⅳ的在正负离子模式下的准分子离子峰
2. 7 线性关系考察 吸取黄芪皂苷Ⅰ对照品溶液1(0. 022 6 mg/mL)0. 75、1、1. 5 μL以及黄芪皂苷Ⅰ对照品溶液2(0. 002 26 mg/mL)1、2. 5、4 μL;黄芪皂苷Ⅱ对照品溶液1(0. 002 30 mg/mL)2、3、4、5 μL以及黄芪皂苷Ⅱ对照品溶液2(0. 001 15 mg/mL)0. 5、1. 5、3 μL;黄芪皂苷Ⅳ对照品溶液3(0. 000 196 mg/mL)1、2、3 μL,黄芪皂苷Ⅳ对照品溶液2 (0. 000 980 mg/mL)1. 5、3、5 μL黄芪皂苷Ⅳ对照品溶液1(0. 009 80 mg/mL)0. 75、1、1. 5、2 μL分别应用UPLC-QTOF/MS进行检测,获得信号强度(及色谱峰峰面积),绘制标准曲线,计算回归方程。见下表。表明黄芪皂苷Ⅰ在0. 002 26 μg~0. 033 9 μg内线性关系良好。黄芪皂苷Ⅱ在0. 000 575 μg~0. 011 5 μg内线性关系良好。黄芪皂苷Ⅳ分别在0. 000 196 μg~0. 002 94 μg以及0. 002 94 μg~0. 019 6 μg内线性关系良好。
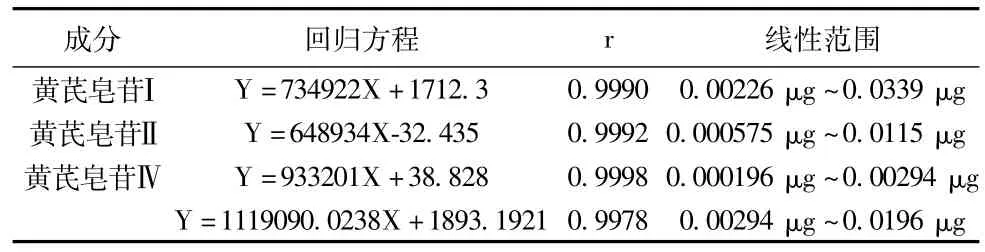
表2 黄芪皂苷Ⅰ、Ⅱ、Ⅳ的线性关系及范围
2. 8 精密度试验 取同一样品,按上述2. 1中供试品溶液制备方法,制备供试品溶液,按2. 5色谱及2. 6质谱条件连续进样6次,测定峰面积。结果表明,黄芪皂苷Ⅰ、黄芪皂苷Ⅱ和黄芪皂苷Ⅳ的峰面积RSD%分别为2. 7%,1. 2%,1. 5%。
2. 9 重复性试验 取同一样品,按上述2. 1中供试品溶液制备方法,平行制备6份供试品溶液。按2. 5色谱2. 6质谱条件分别进样,测定峰面积。结果表明,黄芪皂苷Ⅰ、黄芪皂苷Ⅱ和黄芪皂苷Ⅳ的峰面积RSD%分别为2. 4%,2. 6%,1. 8%。取同一样品,按上述1. 2. 2中供试品溶液制备方法,平行制备6份供试品溶液。按2. 4色谱2. 5质谱条件分别进样,测定峰面积。结果表明,黄芪皂苷Ⅰ、黄芪皂苷Ⅱ的峰面积均为0;黄芪皂苷Ⅳ的峰面积RSD%2. 7%。
2. 10 稳定性试验 取同一样品,按上述2. 1中供试品溶液制备方法,按2. 5色谱及2. 6质谱条件在1,2,4,8,12,24 h分别进样,测定峰面积。结果表明,黄芪皂苷Ⅰ、黄芪皂苷Ⅱ和黄芪皂苷Ⅳ的峰面积RSD%分别为2. 6%,2. 9%,3. 1%。
2. 11 加样回收率试验 取同一样品,每份0. 500 g,精密称定,分别精密加入黄芪皂苷Ⅰ对照品溶液A 12 mL、黄芪皂苷Ⅱ对照品溶液A 30 mL和黄芪皂苷Ⅳ对照品溶液A 0. 9 mL,按上述1. 2中供试品溶液制备方法,按2. 4色谱2. 5质谱条件分别进样,测定峰面积。计算回收率,见表3、表4。

表3 未碱洗供试品黄芪皂苷Ⅰ、Ⅱ、Ⅳ加样回收率结果

表4 碱洗供试品黄芪皂苷Ⅰ、Ⅱ、Ⅳ加样回收率结果
2. 12 样品的含量测定 取8批黄芪样品,按上述2. 1及2. 2中供试品溶液制备方法,制备供试品,按2. 5色谱及2. 6质谱条件项下操作,测定3种成分的含量。
2. 13 数据处理 数据预处理是采用Waters公司的Masslynx 4. 1软件,通过导入离子峰并适当调整积分参数的方式完成色谱峰的识别与峰匹配。两种不同制备方法的供试品溶液样品的分类模式采用多变量的主成分分析法(PCA)以及偏最小二乘法(OPLSDA)进行识别。
3 结果
3. 1 黄芪皂苷Ⅰ、黄芪皂苷Ⅱ和黄芪皂苷Ⅳ的含量测定 在负离子模式下,调整适当的碰撞能,能够在一级质谱图中得到黄芪皂苷Ⅰ、黄芪皂苷Ⅱ和黄芪皂苷Ⅳ稳定的准分子离子峰[M+COOH]-,而正离子模式下以上3种化合物多以碎片离子形式存在(见图2),因此选择在负离子模式下对不同来源黄芪饮片碱洗前后供试品溶液中以上3种化合物进行含量测定。结果如下,见表6。

表5 饮片碱洗前后供试品溶液中黄芪皂苷Ⅰ、黄芪皂苷Ⅱ和黄芪皂苷Ⅳ的含量

图1 正、负离子模式下样品总离子流图

图2 正、负离子模式下黄芪皂苷Ⅰ、黄芪皂苷Ⅱ和黄芪皂苷Ⅳ对照品一级质谱图

图3 正、负离子模式下未碱洗供试品、碱洗供试品PCA得分图

图4 正、负离子模式下未碱洗供试品、碱洗供试品S-plot图

图5 正、负离子模式下黄芪皂苷Ⅰ、黄芪皂苷Ⅱ及黄芪皂苷Ⅳ在碱洗前后供试品中的量变趋势图
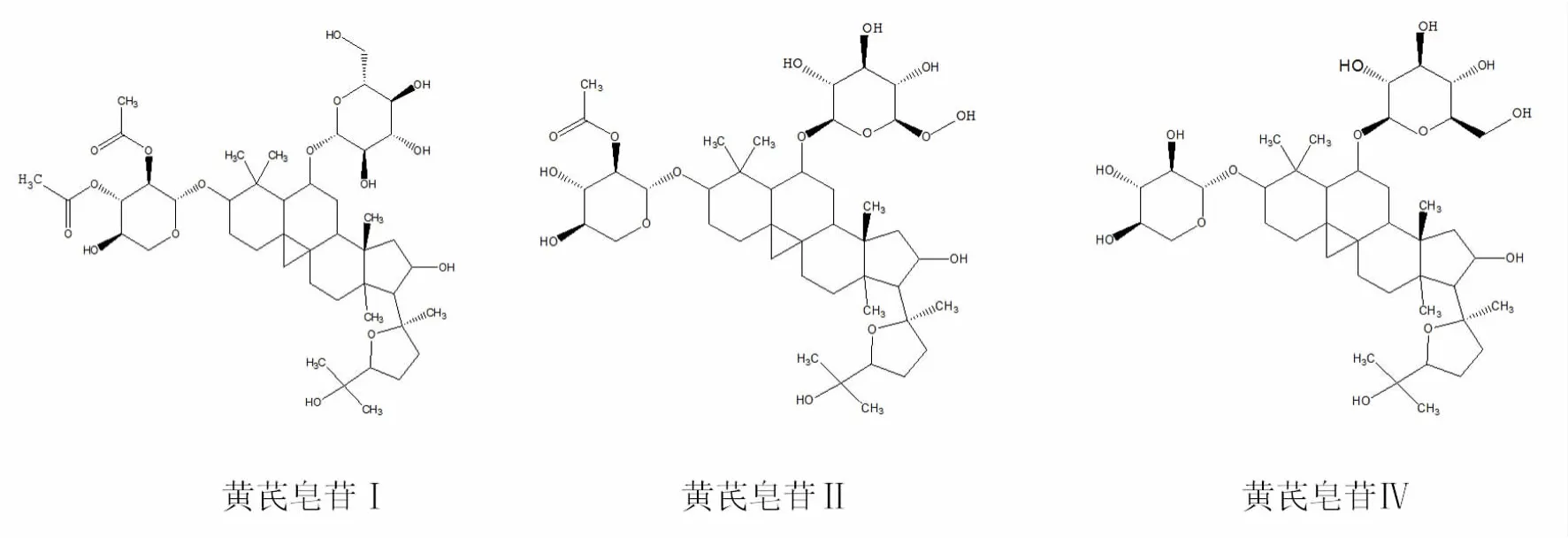
图6 黄芪皂苷Ⅰ、黄芪皂苷Ⅱ和黄芪皂苷Ⅳ结构
4 讨论
4. 1 黄芪饮片碱洗前后供试品PCA与OPLS-DA分析 采用Masslynx 4. 1软件对正、负离子模式下不同来源黄芪饮片碱洗前与碱洗后的供试品色谱图谱中的原始数据进行主成分模式识别分析(PCA)。通过得分图可以看出未碱洗供试品与碱洗供试品能够明显区分开来,且正、负离子模式下分析结果一致(见图3)。尽管来源的不同,黄芪的品质有所差异,氨水洗涤对供试品的影响是较为明显的,8批黄芪饮片的未碱洗供试品与碱洗供试品均能在第一主成分得到良好的区分。说明氨水洗涤在很大程度上改变了黄芪饮片供试品中化学成分群。
为了进一步挖掘2组供试品的变化,再对2组数据进行偏最小二乘模式识别分析(OPLS-DA)。OPLS-DA分析的S-plot见(图4)。图上每个Marker代表一个变量,包含该点的保留时间、质核比以及信号强度3方面信息。横坐标代表Marker对组间差异的贡献程度(协方差)。纵坐标代表Marker对组间差异的贡献程度的可信度(可信度),均在-1~1之间取值。ESI+及ESI-中-1方向均代表未碱洗供试品,1方向代表碱洗供试品。在S点集中离y轴越远的点对组间差异的贡献越大,离x轴越远的点对组间差异贡献的可信度越高。图中可以清晰看出,S点集中,集中在原点以及Y轴的变量较少,说明众多变量在2组间存在较大差异。尤其远离原点的的变量对组间差异的贡献度越高,可信度也越高。量变趋势图(图5)集中了OPLS-DA分析中差异显著的变量,即S-plot图中远离原点的的变量。清晰显示了碱洗步骤在显著降低供试品中黄芪皂苷Ⅰ和黄芪皂苷Ⅱ含量的同时提高黄芪皂苷Ⅳ的含量。
4. 2 黄芪皂苷Ⅰ、黄芪皂苷Ⅱ和黄芪皂苷Ⅳ在碱洗前后供试品中量变关系探究 黄芪皂苷为环菠萝蜜烷型三萜皂苷,苷元多为环黄芪醇。黄芪皂苷Ⅰ、黄芪皂苷Ⅱ和黄芪皂苷Ⅳ结构相近,具有相同的苷元,都在苷元的3位连有一分子木糖,6位连有一分子葡萄糖。区别在于黄芪皂苷Ⅰ的木糖2,3位各有一个乙酰基取代,黄芪皂苷Ⅱ的木糖2位有一个乙酰基取代,黄芪皂苷Ⅳ的木糖无取代基。乙酰基取代在碱性条件下不稳定,容易水解脱酰基[6]。
碱洗供试品制备过程中,碱洗步骤能够使黄芪皂苷Ⅰ、黄芪皂苷Ⅱ脱去酰基转化为黄芪皂苷Ⅳ。含量测定结果显示,碱洗供试品中黄芪皂苷Ⅰ、黄芪皂苷Ⅱ含量为0,黄芪皂苷Ⅳ含量与未碱洗供试品相比显著增加近10倍(见表5)。说明碱洗供试品黄芪皂苷Ⅰ、黄芪皂苷Ⅱ均已水解变构,大部分转化为黄芪皂苷Ⅳ。但是,经比较,碱洗供试品中黄芪皂苷Ⅳ含量仅占未碱洗供试品中黄芪皂苷Ⅰ、Ⅱ、Ⅳ3种成分总和的60%~80%(见表5)。同时分析加样回收率实验结果(见表4),实测值以测得黄芪皂苷Ⅳ的含量为计,供试品所含被测成分含量以样品中所含黄芪皂苷Ⅰ、Ⅱ、Ⅳ的总量为计,加入对照品量以加入的黄芪皂苷Ⅰ、Ⅱ、Ⅳ总量为计,计算加样回收率,在43%~63%之间,RSD%值为17. 69%。表明复杂的供试品制备方法不可避免的增大了系统误差,也影响方法的稳定性及准确性,进而使测量值远离真实值。
综上所述,碱洗步骤能够使黄芪供试品中酰基化的黄芪皂苷如黄芪皂苷Ⅰ、黄芪皂苷Ⅱ脱去酰基转化为黄芪皂苷Ⅳ,显著增加黄芪皂苷Ⅳ的含量。但是该方法实验步骤较为繁琐,系统误差较大,测得黄芪皂苷Ⅳ含量不具代表性。
超高效液相色谱-四级杆-飞行时间串联质谱仪是高分辨串联质谱,其显著特点是高灵敏度、高选择性,含量检测限是紫外检测器的千分之一。因此应用UPLC/Q-TOF-MS技术,按照1. 2. 1供试品制备方法——未碱洗供试品制备方法,制备黄芪饮片供试品,进行黄芪皂苷Ⅰ、黄芪皂苷Ⅱ、黄芪皂苷Ⅳ的含量测定,能够快速准确地测得该三种化合物在饮片中的含量。方法简便、快速、重现性好。
本实验希望能为以黄芪皂苷类成分作为指标成分进行黄芪饮片(或含有黄芪的中成药)质量研究提供实验依据。
参考文献
[1]国家中医药管理局《中华本草》编委会.中华本草[M].上海:上海科学技术出版社,1999:341-344.
[2]黄兆胜.中药学[M].北京:人民卫生出版社,2002:428-429.
[3]国家药典委员会.中华人们共和国药典一部2010年版[S].北京:中国医药科技出版社,2010:283.
[4]LiL,LuoGA,LiangQL,et al. Rapid qualitative and quantitative analyses of Asian ginseng in adulterated American ginseng preparations byUPLC/Q-TOF-MS[J]. JPharmBiomedAnal,2010,52(1):66-72.
[5]Ma ZC,Zhou SS,Liang QD,et al. UPLC-TOF/MS based chemical profiling approach to evaluate toxicity-attenuated chemical composition in combinationofginseng and Radix Aconiti Praeparata[J]. Yao Xue Xue Bao,2011,46(12):1488-1492.
[6]匡海学.中药化学[M].北京:中国中医药出版社,2003:233.
[7]杨琪伟,杨莉,熊爱珍,等.赤芍和白芍抗炎作用的UPLC-MS代谢组学初步研究[J].中国中药杂志,2011,36(6):694-697.
[8]芮雯,冯毅凡,石忠峰,等.黄芪及其蜜炙品的UPLC/Q-TOF-MS分析[J].广东药学院学报,2011,28(1):1-4.
[9]芮雯,冯毅凡,石忠峰,等.不同产地黄芪药材的UPLC/Q-TOFMS[J].指纹图谱研究药物分析杂志,2012,32(4):607-611.
[10]刘颖坤,喻卫武,白岩. UPLC-ESI-Q-TOF-MS在分析山茱萸化学成分中的应用[J].中国现代应用药学,2011,28(3):226-230.
[11]孟宪生,康廷国,叶挺祥,等.中药细辛与藜芦配伍化学成分变化的UPLC/Q-TOF-MS研究[J].中华中医药学刊,2010,28(4):754-759.
[12]杨亮,宇光,梁乾德,等.基于UPLC/Q-TOF-MS/MS不同比例人参配伍藜芦增毒的物质基础及动物毒性关联性研究[J].质谱学报,2012,33(5):257-264.
[13]周思思,马增春,梁乾德,等.基于UPLC/Q-TOF-MS/MS分析附子半夏配伍相反的物质基础[J].化学学报,2012,20(3):284-290.
[14]董林毅,张韻慧,郭婷婷,等. UPLC/Q-TOF-MS/MS测定治咳川贝枇杷滴丸中6种有效成分的含量[J].中国药学杂志,2012,47 (10):230-233.
[15]韩亮,石忠峰,林华庆. UPLC/Q-TOF-MS/MS法分析厚朴化学成分[J].中成药,2013,35(4):366-369.
(2015 -05 -26收稿 责任编辑:张文婷)
中图分类号:R284. 1
文献标识码:A
doi:10. 3969/j. issn. 1673 -7202. 2016. 03. 040
通信作者:曾祖平,女,硕士,主任药师,副教授,硕士生导师,研究方向:中药质量标准及新剂型研究,Tet:(010)52176919,E-mail:zzp600@sohu. com
基金项目:北京市科委“十病十药”研发(编号:Z141100002214015)
