低碳烷烃深加工制烯烃技术的研究进展
2016-03-21张海娟张浩楠奚兆毅
张海娟,高 杰,张浩楠,万 海,奚兆毅
(1. 辽宁石油化工大学 化学化工与环境学部,辽宁 抚顺 113001;2. 中国石油 抚顺石化公司石油三厂,辽宁 抚顺 113001)
技术动态
低碳烷烃深加工制烯烃技术的研究进展
张海娟1,高 杰1,张浩楠1,万 海2,奚兆毅1
(1. 辽宁石油化工大学 化学化工与环境学部,辽宁 抚顺 113001;2. 中国石油 抚顺石化公司石油三厂,辽宁 抚顺 113001)
低碳烷烃深加工脱氢是一种广泛应用的大规模生产纯烯烃的方式。主要综述了低碳烷烃深加工脱氢制烯烃技术的特点及市场份额较高的两种代表性脱氢工艺;详细评述了国内外低碳烷烃深加工脱氢技术现状和商业化的脱氢催化剂体系;总结了丙烷脱氢和异丁烷脱氢的区别和联系;展望了这一技术未来的发展趋势。未来具有经济吸引力的低碳烷烃深加工脱氢技术是最佳的催化剂设计与反应器工程之间的良好的协同作用,而国内目前开发这一技术的首要任务是完成脱氢催化剂的国产化,率先在引进技术的装置上实现脱氢催化剂的替代。
低碳烷烃脱氢;烯烃;脱氢工艺;脱氢催化剂
目前,北美页岩气的开发导致天然气价格大幅下降[1],同时液化石油气用作民用燃料很难为炼厂带来更高效益,寻找液化石油气高附加值利用的途径、充分利用低碳烷烃(丙烷、异丁烷和正丁烷)逐渐受到人们的关注。此外,丙烯和异丁烯下游产品的不断开发利用,也造成了全球性丙烯和异丁烯资源的匮乏。因此,采用低碳烷烃脱氢制烯烃技术拓展低碳烯烃来源、完成液化气高附加值利用,蕴藏着良好的发展机遇和巨大的市场潜力。在20世纪七、八十年代,人们主要关注正丁烷-丁烯-丁二烯的脱氢反应,然而随着市场及应用规模的变化,现在丙烷和异丁烷脱氢已变得越来越重要[2]。低碳烷烃脱氢可以从廉价及可利用的低成本饱和烃原料中获得烯烃,是一种具有相当大工业化影响的方法[3]。
当前低碳烷烃深加工脱氢制烯烃的途径主要有无氧脱氢(催化脱氢,泛指脱氢)、氧化脱氢和膜反应器脱氢[4-6]。氧化脱氢技术由于烷烃及脱氢产物易被深度氧化,距离工业化较远;膜反应器技术也存在渗透压降与反应床层压降合理分配、传质和传热等问题,限制其商业开发[7-8]。目前,只有国外一些公司开发的无氧脱氢技术比较成熟,已实现工业化,而国内尚无自主开发的工业化技术应用。因此,开展具有自主知识产权的无氧脱氢技术研究,尽快实现工业化,具有重大的意义。
本文综述了低碳烷烃深加工脱氢制烯烃技术的特点及市场份额较高的两种代表性脱氢工艺;详细评述了国内外低碳烷烃深加工脱氢技术现状和商业化的脱氢催化体系;总结了丙烷脱氢和异丁烷脱氢的区别和联系。
1 低碳烷烃深加工脱氢制烯烃工艺
低碳烷烃脱氢是一种吸热、平衡控制的反应,需要较高的温度和较低的压力以获得显著的烯烃收率[9]。平衡转化率受热力学限制,需要依靠提高反应温度提高烷烃转化率。这些特性对发展商业过程造成了一定的限制。为了达到经济合理的单程转化率(未反应烷烃分离成本高),反应温度超过550 ℃;而且,烷烃碳数越少,需要的温度越高,相关的工程问题越复杂[9]。但反应温度的升高,有利于较轻烷烃热裂解反应,加速焦炭的生成,造成催化剂失活,从而影响催化剂的选择性和稳定性。为克服这些问题,研发了很多技术。例如,Lummus公司的Catofin技术就是在负压下操作,通过降低反应器压力提高平衡转化率;UOP公司的Oleflex技术在初级阶段也曾尝试采用负压操作来提高平衡转化率,但负压操作在工程操作方面大幅增加了能耗,进而增加了操作成本,Oleflex技术现采用微正压操作[10-11]。
1.1 国外低碳烷烃脱氢5种工艺
自从Frey等[12-13]发现Cr基脱氢催化剂,许多企业为开发商业化脱氢工艺技术进行了大量的研究。低碳烷烃脱氢工艺在操作方面最主要的技术挑战是:1)反应器需充分的供热;2)控制温度,减少裂解产物,最大化转化率;3)催化剂的再生。不同公司有不同的解决方案,由此也诞生了不同的脱氢技术。
世界上低碳烷烃脱氢制烯烃专利技术有:UOP公司的Oleflex工艺、ABB Lummus公司的Catofin工艺、Uhde公司的Star工艺、Snamprogetti/Yarsintz公司的FBD-4工艺以及林德/巴斯夫公司的PDH工艺[9,14]。其中,市场占有率较高的是UOP公司的Oleflex工艺和ABB Lummus公司的Catofin工艺。Oleflex工艺和Catofin工艺已成为丙烷脱氢的代表工艺,而异丁烷脱氢的代表工艺则是Oleflex工艺、Catofin工艺和FBD工艺,林德/巴斯夫公司的PDH工艺只应用于一家示范工厂。
Catofin工艺采用一系列绝热固定床反应器,脱氢和再生周期快速交替,循环操作。FBD工艺使用一个流化床反应器、一个与之连接的再生器,主要在俄罗斯使用,并且规模较小。Oleflex工艺则是由4个串联移动床反应器和一个再生器组成,该工艺结合了两种已成熟的工业化技术,即长链烷烃脱氢的Pacol工艺与Pt重整工艺中的催化剂连续再生技术。而Star和PDH工艺则是采用列管式固定床反应器,切换式再生。这些工艺和反应器设计细节见于诸多文献报道[15-18]。
在采用的催化剂方面,各工艺也有所不同。Oleflex和Star工艺采用的是Pt贵金属催化剂;Catofin,FBD,PDH工艺采用的是Cr-A1催化剂。但PDH工艺的第二代催化剂也已从Cr-A1催化体系改为贵金属催化体系。
1.2 Oleflex工艺与Catofin工艺
1.2.1 Oleflex工艺
Oleflex工艺是UOP公司的烷烃脱氢专利技术,使用Pt基催化剂,利用低碳烷烃作原料,氢气为稀释剂,用以抑制结焦、抑制热裂解和作载热体维持脱氢反应温度,微正压操作,压力0.2~0.5 MPa。该技术的优点是烯烃收率稳定、催化剂再生方法理想、催化剂使用寿命长、装填量少,但移动床技术复杂,投资和动力消耗较高,对原料杂质要求较苛刻[9,14,16]。近年来,使用Pt-Sn催化剂的Oleflex工艺技术已成为脱氢技术的主导。Oleflex工艺流程见图1。
Oleflex工艺脱氢制烯烃技术自1990年在泰国实现工业化以来,一直在持续不断地改进。工艺方面,主要是优化设计、降低投资和减少操作费用。通过操作条件和设计的优化来提高工艺收率,重点集中于提高操作空速、减小反应器尺寸、降低待再生催化剂的焦含量。丙烷脱氢装置规模也不断提高,工业化初期的规模为100 kt/a左右,20世纪末期达到250 kt/a,到21世纪初期进一步提高至300~350 kt/a,从2004年开始一些400 kt/a以上的大型丙烷脱氢装置开始建设。目前,中国公司引进的UOP公司的Oleflex工艺技术,丙烷脱氢装置生产规模达到了600 kt/a[19]。

图1 Oleflex工艺流程(反应和再生)Fig. 1 UOP Oleflex process(reaction section and regeneration section).CCR:continuous catalyst regeneration;SHP:selective hydrogenation process.
催化剂方面,则是不断开发新一代催化剂,主要突破点为调整Pt/Sn比,降低Pt含量,提高Pt的利用率。目前己有DeH-6,DeH-8,DeH-10,DeH-12,DeH-14,DeH-16六代催化剂工业化。第一代催化剂为DeH-6Pt催化剂。1996年开发了第四代催化剂DeH-12,丙烷单程转化率35%~40%,生成丙稀的选择性为89%~91%。2003年位于西班牙的350 kt/a装置已使用第五代催化剂DeH-14。目前,第六代催化剂DeH-16已用在中国投资兴建的福建美德和烟台万华的丙烷脱氢装置上,Pt含量降到0.3%(w)。
近三年来美国UOP公司已向中国19家生产商转让了Oleflex丙烷脱氢技术。自这项技术在1990年首次工业应用以来,UOP公司已交付了多套C3Oleflex和C4Oleflex工业装置。近期,UOP公司在中国张家港新建催化剂生产基地,第一阶段投资的主要产品就是丙烷脱氢工艺所用的Oleflex催化剂。
1.2.2 Catofin工艺
Catofin工艺的开发是基于20世纪40年代Houdry公司开发的丁烷生产丁二烯的Catadiene技术,采用Cr基催化剂。Catofin工艺是ABB Lummus公司开发的C3~C5烷烃脱氢生产单烯烃技术,可由丙烷脱氢制丙烯、异丁烷脱氢制异丁烯、丁烷脱氢制正丁烯和丁二烯。Catofin工艺流程见图2[9,14,20]。

图2 Catofin工艺流程Fig.2 Catofin process flow diagram.PSA:pressure swing adsorption.
Catofin工艺分为4个工段:脱氢制烯烃、反应器排放料的压缩、产品的回收和精制。在一个全循环中,要进行烃蒸气脱氢,反应器内用蒸汽清洗、空气吹扫、预热催化剂并烧掉少量沉积在催化剂上的结焦(基于催化剂的质量分数低于0.1),然后抽真空、复原,开始另一次循环,约23 min切换一次。为了连续生产,Catofin工艺通常采用5台反应器,其中2台生产、2台催化剂再生、1台吹扫[9,20]。
Catofin工艺技术的主要特点是:采用逆流流动循环固定床反应器,在反应器中空气向下、烃类向上流动;使用非贵金属催化剂,对原材料杂质要求低,价格便宜,催化剂寿命为3年,且无催化剂损失。反应器在负压条件下操作,反应中没有氢的再循环,没有蒸汽稀释,装置运行稳定。但是,负压操作需要不停地使用氮气进行置换,导致这一工艺技术中氮气的消耗非常大,而且由于催化剂单程运转的时间非常短(约23 min),需要不停地切换。因此,Catofin工艺相对于Oleflex工艺而言,在降低能源消耗、经营成本和操作费用上并不占优势。Catofin工艺也在不停地进行改进,反应物的入口温度已经从最初的650 ℃降至590 ℃,可有效提高烯烃总收率。
1.3 国内低碳烷烃脱氢技术现状
在我国,国产化的低碳烷烃深加工脱氢制烯烃技术还没有商业应用。近二十多年来,国内有多家科研院所、高校及一些公司进行了丙烷和异丁烷脱氢的研究。中国石化的上海石化研究院、石油科学研究院和抚顺石油化工研究院的研究较为深入,陆续编制了脱氢工艺包,并完成了工艺包的审查[21]。据最新报道,中国石油大学(华东)成功开发非贵金属氧化物脱氢催化剂的丙烷/丁烷脱氢技术,在山东恒源工业试验取得了成功。而由兰州物理研究所开发的Al2O3负载Cr2O3型异丁烷脱氢催化剂,在东营佳昊化工进行了工业试验。
面对低碳烷烃脱氢市场的蓬勃发展态势,国内一些企业采用了引进技术策略。近几年来,国内陆续引进了多套丙烷脱氢和异丁烷脱氢装置,单丙烷脱氢总产能就超过10 Mt/a。天津渤化公司的600 kt/a丙烷脱氢、宁波海越600 kt/a丙烷脱氢、烟台万华600 kt/a丙烷脱氢制丙烯及卫星石化450 kt/a丙烷脱氢等装置相继投产,生产出合格的丙烯产品。丙烷脱氢和异丁烷脱氢大幕在中国彻底拉开,成为石化行业最耀眼的投资项目之一,多个项目预计于未来两三年内投产。相对于丙烷脱氢而言,引进和投产的异丁烷脱氢装置较多,并且多集中于山东省,装置规模主要在100~300 kt/a。目前,国内已经投产的丙烷脱氢、异丁烷脱氢装置见表1。
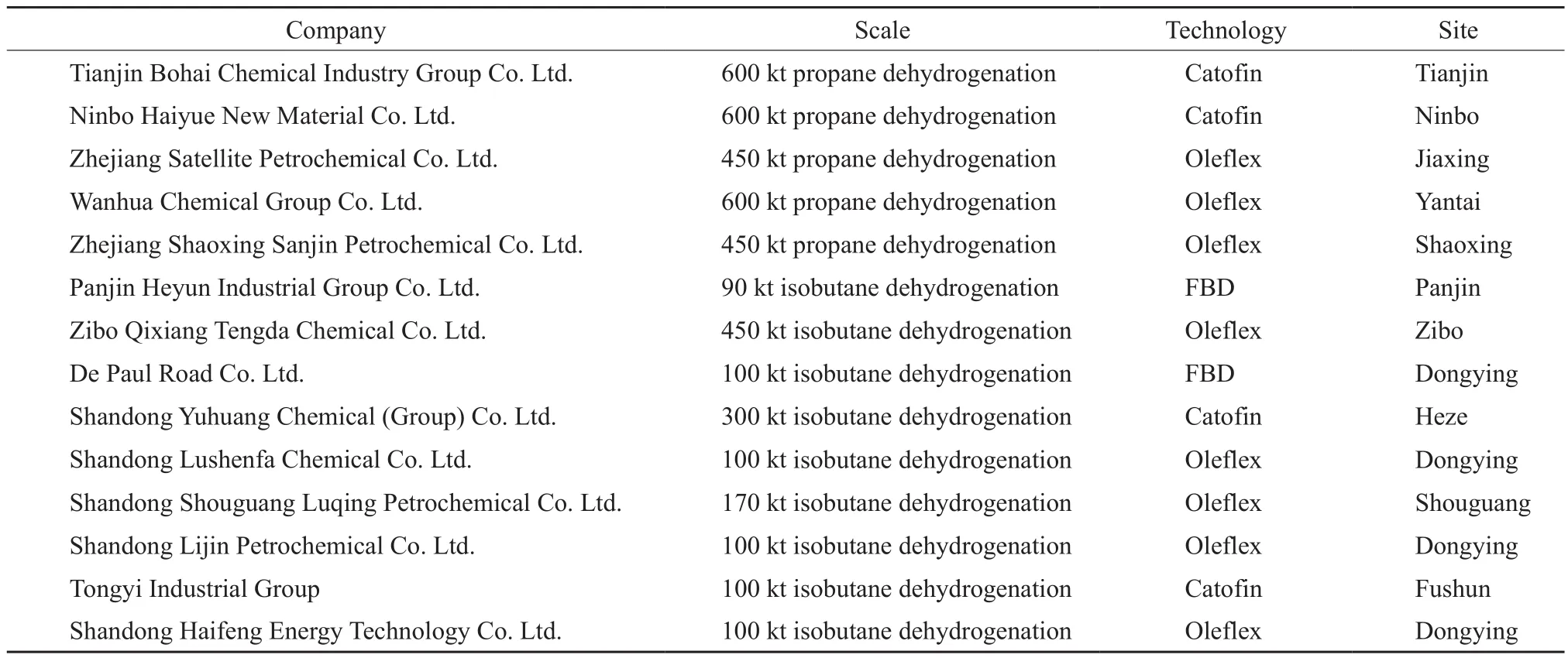
表1 国内已经投产低碳烷烃脱氢装置Table 1 Domestic light paraffin dehydrogenation units which have put into production
2 低碳烷烃脱氢催化体系
低碳烷烃脱氢催化剂主要有两类。一类是Pt基贵金属系列催化剂,以Al2O3、铝酸锌或镁铝尖晶石做载体,Pt为主活性组分,Sn为主要助剂,如UOP开发的DH系列催化剂;另一类是Cr基氧化物系列催化剂,主要是Cr2O3/Al2O3催化剂,如Lummus公司的Cr基催化剂[22]。Cr2O3/γ -Al2O3催化剂对低碳烷烃的脱氢具有良好的活性,该催化剂对原料中杂质的要求比较低,与贵金属催化剂相比,价格便宜,但是此类催化剂容易积碳失活。贵金属Pt催化剂的主要优点在于活性高,选择性较好,但是成本较高。Fe基催化剂虽然在乙苯脱氢中有很好的活性,但是在低碳烷烃脱氢反应中基本无活性[9]。商业化的低碳烷烃脱氢催化剂主要是Pt基催化剂和Cr基催化剂。
2.1 Pt基贵金属催化剂
目前,制约Pt基催化剂广泛应用的主要因素是其高成本和由积碳造成的催化剂失活。Pt基催化剂的研究主要集中在选择合适的载体和助剂,以提高催化剂的性能,降低成本。国内外的公司和企业将丙烷脱氢催化剂的研究重点集中在活性组分、载体及助剂等方面的改进提高。其中,开发新型载体、添加新型助剂或对传统的γ -Al2O3负载的贵金属进行改进研究最为活跃[23-32]。
很多研究者尝试使用大比表面积碳材料或介孔分子筛来实现活性组分的良好分散,提高催化剂脱氢性能。也有人尝试从载体的酸碱性、Sn的加入方式、Pt/Sn金属比例、Pt金属粒径的影响等方面进行研究,期望提高催化剂性能[32-37]。
Pt-Sn催化体系相互作用的模型如图3所示[9]。载体的比表面积、Pt和Sn负载量、Pt/Sn比和还原温度都影响着Pt-Sn之间的相互作用。载体比表面积减小、Pt和Sn负载量增加、Pt/Sn比降低或还原温度过高,都会造成活性金属与助剂状态发生图3中从左至右的变化,产生Pt-Sn合金,从而使催化剂失活。因此,在Pt-Sn催化体系中,催化剂载体比表面积要大、Pt和Sn负载量及Pt/Sn比例适当,还原温度不宜过高。

图3 Pt-Sn相互作用模型Fig.3 Models of Pt-Sn interaction.
目前,商业化的贵金属催化剂无法从本质上解决低碳烷烃深加工脱氢技术所遇到的瓶颈,即高烷烃单程转化率、烯烃选择性与催化剂积碳失活的矛盾。其根本的原因就是用于脱氢的Pt基催化剂与应用于其他领域的传统贵金属Pt基催化剂在Pt颗粒的分散度、粒径分布及表面形貌等微观结构上有着明显的区别。如何合理设计催化剂织构、匹配活性组分与助剂是获得高效、低积碳率的低碳烷烃Pt基催化剂的关键,而这方面的研究相对较少。
Pt基贵金属脱氢催化剂失活的主要原因就是积碳。张海娟等[38]考察了低碳烷烃Pt基贵金属催化剂的失活过程,确定了影响催化剂结焦的主要因素。对焦的芳香性质和石墨化程度来说,温度是主要的影响因素,氢烃比次之。Afonso等[39]认为Pt基贵金属催化剂表面积碳的形成过程主要包括以下几个步骤:1)烃类的连续脱氢/环化;2)直链烃的聚合;3)Diels-Alder类反应。催化剂表面的酸量越多,越容易促进裂解、异构化等副反应的发生,增加催化剂表面的积碳量。积碳前体主要是通过金属表面的作用(脱氢)和载体表面的作用(裂解)产生的。
李庆[40]推测了Pt基催化剂上丙烷脱氢催化剂的结焦机理,其示意图见图4。在丙烷脱氢制丙烯过程中,丙烷首先被吸附在催化剂的活性位上,脱氢生成炭的前体。该前体一部分生成覆盖活性位的焦炭,另一部分可迁移至载体,参与载体上焦炭的生成。此外,生成的丙烯可直接吸附在载体的酸性位上,参与载体上的结焦过程。而在催化剂活性位上生成的焦多为不定型焦,其氢碳比相对较高;在载体上形成的焦多为石墨型焦,其氢碳比相对较低。且随反应的进行,石墨型焦的数量大幅增长,而不定型焦的增长速度则较为缓慢。
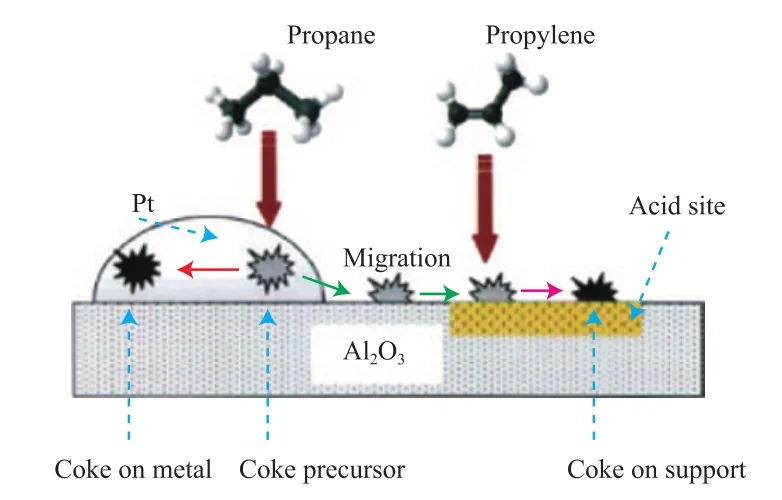
图4 丙烷脱氢催化剂结焦机理的示意图Fig.4 Coking mechanism on the Pt catalyst for propane dehydrogenation.
比较典型的低碳烷烃脱氢贵金属催化剂为Oleflex工艺所采用的Pt-Sn催化剂,其发展情况见表2[16]。由表2可见,在DeH-10催化剂之后,新一代催化剂的改进主要集中于降低Pt含量、调整Sn助剂和碱金属助剂的含量,以期活性组分与助剂之间达到最佳匹配,提高催化剂的活性和抗结焦性能。

表2 UOP 公司Oleflex工艺催化剂发展情况Table 2 Development of the dehydrogenation catalysts of the UOP Oleflex process
未来,Pt基贵金属脱氢催化剂的设计应从活性金属与助剂协同作用和可控的调节活性中心原子簇晶粒大小为突破点,达到提高催化剂的寿命和抗结焦性能的目的。通过调节载体表面性质,使活性组分更多地锚定于助剂界面,从而形成更多的有效活性位;对Pt颗粒进行可控制备,避免生成对C—C键活化效果好、有利于裂解反应的小的Pt颗粒;控制合成一定粒径的Pt颗粒,使其对C—H键活化效果好,利于脱氢反应。所设计的脱氢催化剂应具有良好的目的产物选择性、稳定性和抗结焦性能。
2.2 Cr基氧化物催化剂
由于Frey和Huppke的开创性工作,负载型Cr2O3催化剂在烷烃脱氢反应中的优越性能广为所知,并且Cr2O3/Al2O3催化剂成为商业催化剂的广泛选择[12]。Frey和Huppke最初的Cr基脱氢催化剂是通过铝和Cr的硝酸盐和氢氧化铵的共沉淀法制备的,现在更多改良的制备方法已经开发出来[41]。
早期,Poole等[42]详细地总结了Cr2O3/γ-Al2O3催化剂的主要性能。随后,许多学者研究了CrOx/ SiO2,CrOx/Al2O3,CrOx/ZrO2催化剂[43-46]。研究主要集中在:1)活性位的识别;2)对Cr2O3形态的制备和处理条件以及催化剂活性、选择性的影响;3)脱氢机理研究。
Cr系氧化物催化剂的活性和稳定性受活性位点与环境的相互作用的影响很大。活性物种的氧化状态已成为许多年来讨论和争议的话题。很多学者认为Cr系氧化物催化剂脱氢的活性中心是配位不饱和的Cr3+,关于何种类型的Cr3+是脱氢活性中心没有一致的结论。以Puurunen等[47]为代表的学者认为非还原Cr3+和Cr3+簇的催化活性比还原Cr3+和独立的Cr3+要高。而以Derossi等[45]和Hakuli等[48]为代表的学者提出单核还原Cr3+是活性中心。但是也有部分学者提出Cr2+为活性物种。Gorriz等[49]研究认为,Cr3+、Cr5+和Cr6+形成多种化合物,并具有不同的还原性和催化行为。Cr5+物种和催化剂的初活性相关,但主活性中心是Cr2+,而丙烯的选择性主要由Cr3+物种决定。
目前,越来越多的学者开始大量研究Cr系催化剂上烷烃脱氢机理。但是由于反应条件、催化剂测试手段不同,关于烷烃脱氢反应的活化、决速步骤等方面存在着很大的差异[48]。目前比较一致的看法是,烷烃脱氢反应在Cr催化剂上主要由3个步骤来完成,如图5所示[44]。由图5可知,首先烷烃在不饱和的Crn+中心吸附,Crn+可以是孤立的或聚集的(Crn+簇团);其次C—H键断裂,O—H键和Cr—C键形成,最后在催化剂表面形成丙烯;最后丙烯从活性中心表面释放,并且生成氢气,催化剂活性位复原。低碳烷烃脱氢的脱氢机理研究,是低碳烷烃转化利用理论研究的基础,对低碳烷烃转化利用具有重要的指导意义。

图5 Cr2O3/Al2O3催化剂烷烃脱氢的反应机理Fig.5 Mechanism for the alkane dehydrogenation over the Cr2O3/Al2O3catalyst.
商业化的Cr2O3/Al2O3催化剂面临最主要的问题就是催化剂失活。Cr2O3/Al2O3催化剂的失活可能由以下原因造成[48]:1)Cr活性中心烧结;2)活性中心被积碳覆盖,阻碍了脱氢反应的进行;3)与Al2O3结合形成不具有催化活性的Cr3+物种,这是催化剂永久失活的主要原因。图6为Cr2O3-Al2O3作用失活模型[9]。由图6可以看出,焙烧使孤立的Cr3+离子形成Cr3+簇团,Al2O3载体的烧结诱捕Cr3+离子进入载体中,与Al2O3的Al3+空位结合,形成一种新的稳定的α-Cr2O3-Al2O3型尖晶石结构,从而丧失了催化活性[22]。这是由于Cr3+和Al3+具有相似的离子半径和电荷数,促进了α-Cr2O3-Al2O3型尖晶石结构的形成。

图6 Cr2O3-Al2O3作用模型Fig.6 Model of the chromia-alumina interaction.
最成功的商业化的Cr系氧化物催化剂就是Catofin工艺所使用的Cr2O3/Al2O3催化剂,由Süd Chemie提供。催化剂以Al2O3为载体,负载Cr2O3,具有良好的机械强度,较强的抗中毒能力,可以耐受烯烃含氧化合物和重金属。Catofin工艺技术商业化催化剂如图7所示。

图7 Catofin工艺应用的Cr2O3/Al2O3催化剂Fig.7 Chromia-alumina catalyst for the Catofin process.
由于Cr系催化剂对原料中杂质的要求比较低,反应活性高且价格低廉,原料易得,与贵金属催化剂相比具有成本优势。但是,Cr系催化剂中Cr是重金属组分,环保治理费用较高,且稳定性差,在反应中催化剂失活较快需要反复频繁再生,工业操作相对麻烦。因此,未来开发抗结焦且低负载量的Cr系催化剂是目前低碳烷烃脱氢技术面临的巨大挑战和亟待解决的问题。
3 丙烷脱氢与异丁烷脱氢技术的比较
烷烃脱氢(CnH2n+2CnH2n+H2)是强吸热反应,单程转化率受热力学平衡限制[50-51]。热力学表明,对于烷烃,碳链越长,脱氢越容易,所需反应温度越低。因此,相比丙烷脱氢,异丁烷脱氢所需温度较低。但是,丙烷和异丁烷脱氢的催化体系和脱氢工艺是类似的。由于脱氢温度较低,异丁烷脱氢催化剂单程运转周期较长,积碳量小。
C4烃类具有较多同分异构体,因此异丁烷脱氢产物比较复杂,而丙烷脱氢烯烃产物比较单一,目的异丁烯的选择性要高于目的产物丙烯的选择性。异丁烷脱氢产物流直接进入甲基叔丁基醚单元,省却了异丁烯分离提纯的过程,而丙烷脱氢产物流则要进行深冷分离,最低分离温度接近-100 ℃,分离费用昂贵。因此从投资的角度看,相同规模的异丁烷脱氢装置的投资要相对低一些。
国外的异丁烷脱氢技术应用多集中在20世纪80年代末期、90年代初期,2000年后期投产的异丁烷脱氢装置鲜有报道,但是随着国内对低碳烷烃高附加值利用的关注和重视,中国投资兴建的异丁烷脱氢装置达十余套。在国外已建的异丁烷脱氢工业装置中,与丙烷脱氢采用的技术相反,Catofin工艺市场占有率略高于Oleflex工艺,而前苏联则多采用FBD工艺,这与时代大环境和那个时期的技术特点有关。在我国的低碳烷烃脱氢市场中,引进的丙烷和异丁烷脱氢技术采用Oleflex工艺较多。
综合比较经济性、能耗及安全环保等因素,Oleflex技术在丙烷脱氢应用市场中占有一定优势。但是对异丁烷脱氢而言,Oleflex技术、Catofin技术及FBD技术分庭抗礼。受低油价的影响,在目前市场中,由于丙烯短缺,丙烷脱氢装置开工率好于异丁烷脱氢装置。
4 低碳烷烃脱氢技术未来发展趋势
目前,低碳烷烃脱氢技术开发所面临的问题主要有两个方面:从催化剂角度讲,降低贵金属和Cr2O3的负载量,同时提高催化剂的活性、稳定性和抗结焦性,延长催化剂的再生周期和获得稳定的再生性能;从工艺工程方面,就是如何实现超低压力的低碳烷烃脱氢工艺,同时降低后期的分离能耗。
传统的催化剂很难在高温、低压、高空速条件下实现催化剂的高稳定性。国际上现采用添加稀释气氢气和负压操作来达到提高催化剂活性和稳定性的目的,这些技术没有从本质上解决催化剂脱氢活性和抗结焦性之间的矛盾。从现有烷烃脱氢技术研究趋势看,高烷烃脱氢活性、低催化剂积碳速率是目前的催化剂研究热点,也是从根本上解决催化剂脱氢活性与抗结焦性之间矛盾的有效方法之一。而未来具有经济吸引力的低碳烷烃深加工脱氢技术应是最佳的催化剂设计与反应工程之间良好的协同。国内开发这一技术的首要任务是完成低碳烷烃脱氢催化剂的国产化,率先在引进技术的装置上实现脱氢催化剂的替代,为尽快实现这一技术国产化迈出坚实的一步。
[1] Tallman M J,Klavers R. North American olefin producers riding the shale gas wave[J]. Hydrocarbon Process,2013,4:37-40.
[2] Carra S,Forni L. Catalytic dehydrogenation of C4hydrocarbons over chromia-alumina[J]. Cat Rev-Sci Eng,1971,5(1):159-198.
[3] Weckhuysen B M,Wachs I E,Schoonheydt R A. Surface chemistry and spectroscopy of chromium in inorganic oxides[J]. Chem Rev,1996,96(8):3327-3350.
[4] Chang Jongsan,Roh Hyunseog,Park Seok Min,et al. Propane dehydrogenation over a hydrogen permselective membrane reactor[J]. Bull Korean Chem Soc,2002,23(5):674-678.
[5] Bhasin M M,McCain J H,Vora B V,et al. Dehydrogenation and oxide hydrogenation of paraf fi ns to ole fi ns[J].Appl Catal,A,2001,221(1/2):397-419.
[6] 张海娟,李江红,张喜文,等. Pt-Sn催化剂上异丁烷催化脱氢反应宏观动力学模型[J].石油化工,2010,39(11) :1228-1231.
[7] Quicker P,Hollein V,Dittmeyer R. Catalytic dehydrogenation of hydrocarbons in palladium composite membrane reactors[J].Catal Today,2000,56(1):21-34.
[8] Miachon S,Dalmon J A. Catalysts in membrane reactors:What about the catalyst?[J].Top Catal,2004,29(1/2):59-65.
[9] Sanfilippo D,Miracca I. Dehydrogenation of paraffins:Synergies between catalyst design and reactor engineering[J].Catal Today,2006,111(1/2):133-139.
[10] 肖锦堂. 烷烃催化脱氢生产C3~C4烯烃工艺(之一) [J].天然气工业,1994,14(2):64-69.
[11] 肖锦堂. 烷烃催化脱氢生产C3~C4烯烃工艺(之二)[J].天然气工业,1994,14(3):69-73.
[12] Frey F E,Huppke W F. Equilibrium dehydrogenation of ethane,propane,and the butanes[J]. Ind Eng Chem,1933,25(1) :54-59.
[13] Phillips Petroleum Co. Processes for converting hydrocarbons:US2098959[P].1937-11-16.
[14] Vora B V. Development of dehydrogenation catalysts and processes[J].Top Catal,2012,55(19/20):1297-1308.
[15] 严乐平. 丙烷脱氢制丙烯生产技术的应用前景[J].上海化工,2010,35(7):22-27.
[16] 张琦,隋志军,顾雄毅,等. 丙烷脱氢分离工艺的模拟与分析[J].石油化工,2015,44(4):421-428.
[17] 周巍. 丙烷脱氢制丙烯技术浅析 [J].石油化工设计,2013,30(3):36-38.
[18] Nawaz Z,Chu Y,Yang W,et al. Study of propane dehydrogenation to propylene in an integrated fluidized bed reactor using Pt-Sn/Al-SAPO-34 novel catalyst[J]. Ind Eng Chem Res,2010,49(10):4614-4619.
[19] 刘淑鹤. 丙烷脱氢反应热力学分析和动力学研究[D].抚顺:辽宁石油化工大学,2009.
[20] 朱义才. 丙烷脱氢制丙烯技术经济分析[J].当代石油石化,2012,212(8):36-42.
[21] 王培超,曹世凌,伍宝州. 丙烷脱氢制丙烯技术的工业应用探讨[J].中外能源,2015,30(5):85-89.
[22] 刘乔,董秀芹,余英哲,等. 丙烷无氧脱氢制丙烯工艺和催化剂的研究进展[J].石油化工,2014,43(6):713-720.
[23] Razavian M,Fatemi S. Synthesis and application of ZSM-5/ SAPO-34 and SAPO-34/ZSM-5 composite systems for propylene yield enhancement in propane dehydrogenation process[J]. Microporous Mesoporous Mater,2015,201:176-189.
[24] Shohreh M K,Razavian F M. Hierarchical SAPO-34 catalytic support for superior selectivity toward propylene in propane dehydrogenation process[J]. Korean J Chem Eng,2015,32(7):1289-1296.
[25] Ren Yingjie,Wang Jie,Hua Weiming,et al. Ga2O3/HZSM-48 for dehydrogenation of propane:Effect of acidity and poregeometry of support[J]. J Ind Eng Chem,2012,18(2):731-736.
[26] Long Liuliu,Lang Wanzhong,Yan Xi,et al. Yttrium-modified alumina as support for trimetallic PtSnIn catalysts with improved catalytic performance in propane dehydrogenation[J]. Fuel Process Technol,2016,146:48-55.
[27] Zhang Yiwei,Zhou Yuming,Huang Li,et al. Structure and catalytic properties of the Zn-modified ZSM-5 supported platinum catalyst for propane dehydrogenation[J].Chem Eng J,2015, 270:352-361.
[28] Biloen P,Dautzenberg F M,Sachtler W M H. Catalytic dehydrogenation of propane to propylene over platinum and platinum-gold alloys[J]. J Catal,1977,50(1):77-86.
[29] Kogan S B,Schramm H,Herskowitz M. Dehydrogenation of propane on modified Pt/θ-alumina:Performance in hydrogen and steam environment[J]. Appl Catal,A,2002,208(1/2):185-191.
[30] Zhou S,Zhou Y,Shi J,et al. Synthesis of Ce-doped mesoporous γ-alumina with enhanced catalytic performance for propane dehydrogenation[J]. J Mater Sci,2015,50(11):3984-3993.
[31] Liu Lei,Deng Qingfang,Liu Yuping,et al. HNO3-activated mesoporous carbon catalyst for direct dehydrogenation of propane to propylene[J].Catal Commun,2011,16(1):81-85.
[32] Kumar S M,Holmen A,Chen De. The influence of pore geometry of Pt containing ZSM-5,Beta and SBA-15 catalysts on dehydrogenation of propane[J].Microporous Mesoporous Mater,2009,126(1/2):152-158.
[33] Zangeneh T F,Taeba A,Gholivandc K,et al. The effect of mixed HCl-KCl competitive adsorbate on Pt adsorptionand catalytic properties of Pt-Sn/Al2O3catalysts in propane dehydrogenation[J]. Appl Surf Sci,2015,357(Part A):172-178.
[34] Liu Jie,Liu Changcheng,Ma Aizeng,et al. Effects of Al2O3phase and Cl component on dehydrogenation of propane[J]. Appl Surf Sci,2016,368:233-240.
[35] Zhang Yiwei,Zhou Yuming,Qiu Anding,et al. Propane dehydrogenation on PtSn/ZSM-5 catalyst:Effect of tin as a promoter[J].Catal Commun,2006,7(11):860-866.
[36] Kumar M S,Chen D,Holmen A,et al. Dehydrogenation of propane over Pt-SBA-15 and Pt-Sn-SBA-15:Effect of Sn on the dispersion of Pt and catalytic behavior[J].Catal Today,2009,142(1/2):17-23.
[37] Caeiro G,Carvalho R H,Wang X,et al. Activation of C2-C4alkanes over acid and bifunctional zeolite catalysts[J]. J Mol Catal A:Chem,2006,255(1):131-158.
[38] 张海娟,王振宇,李江红,等. 反应条件对丙烷脱氢催化剂积炭行为的影响[J],天然气化工,2014,39(2):38-42.
[39] Afonso J C,Aranda D A G,Schmal M,et al. The chemistry of coke deposits formed on a Pt-Sn catalyst during dehydrogenation of n-alkanes to mono-olefins[J]. Fuel Process Technol,1994,41(1):13-25.
[40] 李庆. Pt催化剂上丙烷脱氢反应与结焦动力学[D].上海:华东理工大学,2012.
[41] 谭晓林,马波,张喜文,等. Cr系丙烷脱氢催化剂研究进展[J].化工进展,2010,29(1):51-57.
[42] Poole C P,Maclver D S. The physical-chemical properties of chromia-alumina catalysts[J]. Adv Catal,1967,17:223-314.
[43] Hakuli A. Preparation and characterization of supported CrOxcatalysts for butane dehydrogenation[D]. Finland:Helsinki University of Technology,1999.
[44] Weckhuysen B M,Schoonheydt R A. Alkane dehydrogenation over supported chromium oxide catalysts[J]. Catal Today,1999,51(2):223-232.
[45] Derossi S,Ferraris G,Fremiotti S,et al. Propane dehydrogenation on chromia/silica and chromia/alumina catalysts[J]. J Catal,1994,148(1):36-46.
[46] Cutrufello M G,De Rossi S,Ferino I,et al. Characterisation and activity of chromia-zirconia catalysts for propane dehydrogenation[J].Thermochim Acta,2005,434(1/2):62-68.
[47] Puurunen R L,Weckhuysen B M. Spectroscopic study on the irreversible deactivation of chromia/alumina dehydrogenatio catalysts[J].J Catal,2002,210(2):418-430.
[48] Hakuli A,Kytökivi A,Krause A O I,et al. Initial activity of reduced chromia/alumina catalyst in n-butane dehydrogenation monitored by on-Line FT-IR gas analysis[J].J Catal,1996,161(1):393-400.
[49] Gorriz O F,Cadu L E. Supported chromium oxide catalysts using metal carboxylate complexes:Dehydrogenation of propane[J]. Appl Catal,A,1999,180:247-260.
[50] 张海娟,李江红,王振宇,等. 丙烷脱氢反应的热力学分析[J].石油化工,2012,41(增刊):86-88.
[51] 张海娟,张舒冬,李江红,等. 异丁烷脱氢反应的热力学分析[J].化学通报,2011,74(7):628-632.
(编辑 王 馨)
Progresses in processes and catalysts for dehydrogenation of light paraffins to olefins
Zhang Haijuan1,Gao Jie1,Zhang Haonan1,Wan Hai2,Xi Zhaoyi1
(1.Chemical Engineering and Environmental Engineering,Liaoning Shihua University,Fushun Liaoning 113001,China;
2. Fushun Petrochemical Company Refinery NO.3,CNPC,Fushun Liaoning 113001,China)
The dehydrogenation of light paraffins was widely applied in the large-scale production of olefins. The characteristics of the dehydrogenation technologies were reviewed. Among them,the Oleflex process and Catofin process were illustrated due to their high market share. Two catalyst systems for the dehydrogenation processes were discussed. The difference between the propane dehydrogenation and the iso butane dehydrogenation were summarized. It was predicted that,the favourable synergy between the catalyst design and the reactor engineering would be the development trend of the light paraffin dehydrogenation in future. The most urgent task was the investigation and production of domestic dehydrogenation catalysts.
dehydrogenation of light paraffins;olefins;dehydrogenation process;dehydrogenation catalysts
1000-8144(2016)12-1411-09
TQ 203.2
A
10.3969/j.issn.1000-8144.2016.12.001
2016-08-24;[修改稿日期]2016-09-11。
张海娟(1973—),女,辽宁省葫芦岛市人,博士,副教授,电话 024-56865303,电邮 zhj_w@163.com。
