Cis-AT和Trans-AT聚酮合酶及其特殊功能域研究进展
2016-03-21张博向文胜东北农业大学生命科学学院哈尔滨150030
张博,向文胜(东北农业大学生命科学学院,哈尔滨 150030)
Cis-AT和Trans-AT聚酮合酶及其特殊功能域研究进展
张博,向文胜
(东北农业大学生命科学学院,哈尔滨150030)
摘要:聚酮合酶(Polyketide synthase, PKS)是产生聚酮化合物的模块化复合酶,聚酮合酶能产生数量众多并具有生物活性的聚酮类天然产物。近年来一类与cis-AT聚酮合酶存在较大差异的trans-AT聚酮合酶被广泛研究。文章介绍cis-AT聚酮合酶与trans-AT聚酮合酶区别及trans-AT聚酮合酶基因簇中特殊功能域研究进展,展望利用功能域改造基因簇研究方向,以期为天然产物改造提供新靶点。
关键词:trans-AT聚酮合酶;特殊功能域;结构改造
张博,向文胜. Cis-AT和Trans-AT聚酮合酶及其特殊功能域研究进展[J].东北农业大学学报, 2016, 47(1): 87-92.
Zhang Bo, Xiang Wensheng. Advances in Cis-AT and Trans-AT polyketide synthases and their special domains[J]. Journal of Northeast Agricultural University, 2016, 47(1): 87-92. (in Chinese with English abstract)
聚酮合酶(Polyketide synthases,PKS)是由具有多种功能的小蛋白功能域组合而成的巨大蛋白复合体。PKS能产生数量众多且具有生物活性的聚酮类天然产物,如临床应用的抗生素红霉素(Erythromycin)和抗寄生虫药物阿维霉素(Aver⁃mectin),以及抗癌药物埃博霉素(Epothilone)和变构霉素(Tautomycetin)等。聚酮化合物具有高度复杂的结构和较强药理学活性,虽然由醋酸盐和丙酸盐组成,但其结构多样性和复杂性使传统化学合成方式难以获得聚酮化合物。随着分子生物学技术不断发展,研究者对聚酮化合物生物合成及其机理了解更加深入,关注聚酮化合物中特殊功能团相关功能域研究与发掘,以期通过功能域改造和重组产生具有新功能团甚至新功能的化合物[1]。本文比较Ⅰ型聚酮合酶中cis-AT型(整合AT型)聚酮化合物生物合成基因簇和trans-AT型(游离AT型)聚酮化合物生物合成基因簇,介绍Ⅰ型聚酮合酶中包含的特殊功能域和近期合成生物学研究进展,为聚酮类化合物结构改造和发掘提供理论基础。
1 cis-AT和trans-AT型聚酮合酶简介
Ⅰ型聚酮合酶主要是催化二碳单元线性延伸和可选择性催化酮基基团的还原反应,这种类似于初级代谢中脂肪酸合成的催化反应产生数量繁多、结构多样的天然产物。聚酮化合物形成过程始于酰基转移酶(Acyltransferase, AT)特异性活化简单前体,如丙二酰(Malonyl)辅酶A、甲基丙二酰(Methlymalonyl)辅酶A或其他起始单元[2],将其装载到酰基载体蛋白(Acyl carrier protein, ACP),完成起始模块(Loading module)。在酰基转移酶活化一个酰基单元后,酮基合成酶(Ketosynthase,KS)则催化克莱森缩合反应(Claisen condensation),将之前模块上的酰基单元和经过脱羧反应后用于延伸的二碳单元融合,并将上一模块中酰基载体蛋白上的中间体转移到所在模块中的酰基载体蛋白,实现二碳单元线性延伸。此外,二碳单元延伸过程中,每一个模块还会存在一些酮基还原酶(Ketoreductase, KR)、脱水酶(Dehydratase, DH)、烯醇还原酶(Enoylreductase, ER)功能域催化β位酮基还原反应,导致聚酮化合物在不同位置上酮基还原程度有所不同,产生结构多样性。当聚酮链延伸到特定程度,通常会在硫酯酶(Thioester⁃ase, TE)功能域作用下将硫脂键切割终止聚酮链延伸。对经典Ⅰ型聚酮合酶而言,除起始模块之外,KS-AT-ACP等3个功能域是构成聚酮合酶最小组成单位(见图1)。
对经典Ⅰ型聚酮合酶的认知主要从放线菌中红霉素生物合成聚酮合酶基因簇中归纳[3]。研究者将重点转移到非常规细菌和未认知基因簇上,发现一系列聚酮基因簇和与之相对应的化合物。这些特殊的聚酮基因簇中,每个模块均缺少酰基转移酶功能域,而底物酰基化则由1个游离在聚酮合酶之外的酰基转移酶负责[4-7];因此每一步克莱森缩合反应中装载的大多数均是相同的丙二酰单元(见图1)。为区别这两种不同类型的Ⅰ型聚酮合酶,使用trans- AT PKS和cis-AT PKS分别命名含有游离酰基转移酶的聚酮合酶和含有整合酰基转移酶功能域的聚酮合酶。此外,trans-AT PKS也被称作AT-less PKS[8],表明整个聚酮合酶上的酰基转移酶缺失,但经典的Ⅰ型聚酮合酶则没有名称对应。
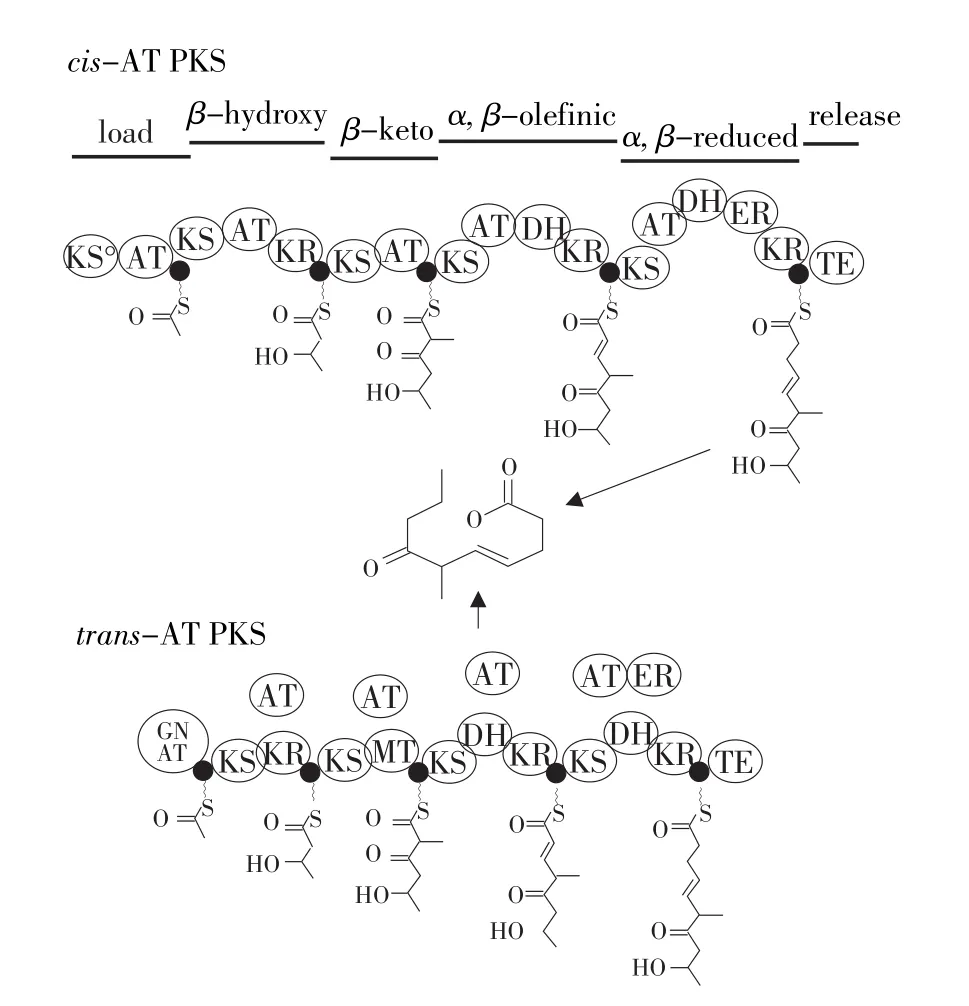
图1 cis-AT聚酮合酶和trans-AT聚酮合酶结构模型及生物合成过程Fig. 1 Biosynthesis of a functional complex polyketide under the action of a cis-AT(textbook) PKS and a trans-AT
首例trans-AT聚酮合酶基因簇是1993年从测序枯草芽孢杆菌168基因组中预测出的bacillaene聚酮合酶基因簇,该菌株并不产生任何聚酮化合物,由于测序技术不成熟导致其序列中存在错误,故无法确认bacillaene聚酮合酶基因簇是功能基因簇抑或是进化生成的残缺基因簇[9]。1999年和2001年,myxovirescins和albicidin生物合成基因簇中单一模块被分别鉴定,展现出与bacillaene聚酮合酶相同的结构特征,即均缺失酰基转移酶功能域[10-11]。事实上,2002年发现的pederin聚酮合酶基因簇是第一个被人们认知的trans-AT聚酮合酶基因簇,首次通过体内试验证明trans-AT聚酮合酶存在,提出游离酰基转移酶为整个聚酮合酶系统提供酰基化底物假设[12]。2003年,发现另外一个trans-AT型聚酮合酶基因簇,即leinamycin生物合成基因簇,通过体外试验验证游离酰基转移酶能提供酰基化底物[8]。随后,trans-AT型聚酮合酶基因簇引起关注,相继发现一系列trans-AT基因簇,如oxazolomycin[13]、virginiamycin M[14]、lankaci⁃dins[15]、Kirromycin[16]等。
2 cis-AT和trans-AT型聚酮合酶区别及特殊功能域
结构上,cis-AT和trans-AT型聚酮合酶唯一区别是酰基转移酶功能域以整合或游离形式存在。系统进化分析表明,两种聚酮合酶系统是从简单脂肪酸生物合成途径按照不同方式独立进化的结果[17];cis-AT主要通过独立模块复制和功能域分化进化形成[18],而trans-AT则多通过不同细菌中水平基因转移而进化形成[19]。由于进化途径不同,大多数trans-AT型聚酮合酶具有一定特性。主要体现在cis-AT型聚酮合酶大多只存在8种功能域组合方式,而在trans-AT型聚酮合酶中则存在超过50余种排列多样和功能特异的功能域模块,大多不遵循经典聚酮合酶生物合成线性规律。
2.1甲基基团添加
大部分cis-AT型聚酮合酶从放线菌中被分离鉴定,由于放线菌能够代谢产生各种类型底物,使大多数cis-AT型聚酮合酶可利用内部整合的酰基转移酶功能域识别特殊底物(例如甲基丙二酰辅酶A)引入α位置上的甲基、乙基等支链[20]。而trans-AT型聚酮合酶主要从一些非放线菌中分离,由于进化方式不同,游离在聚酮合酶之外的酰基转移酶不能直接酰基化含有甲基或乙基支链的丙二酰辅酶A,而只能通过一个S-腺苷甲硫氨酸(SAM)依赖的甲基转移酶引入α位置上的甲基。因此,几乎所有含有甲基侧链的trans- AT型聚酮合酶基因簇中均包含一个甲基转移酶功能域(MT)用于引入与之相对应的甲基基团。
2.2游离AT特殊结构
trans-AT聚酮合酶生物基因簇中酰基转移酶在整个聚酮合酶外均以游离形式游离存在,单一酰基转移酶能重复作用酰基化所有底物。除oxa⁃zolomycin[13]和kirromycin[16]等特例外,所有trans-AT聚酮合酶均利用丙二酰辅酶A为底物进行二碳单元延伸。因此,在trans-AT型聚酮合酶基因簇中含有一个或多个拷贝的AT基因,这些AT基因可融合以二聚体形式存在。此外,某些AT基因可融合一个氧化还原酶功能域对特定位置进行烯醇还原反应,将双键还原为单键,这一氧化还原功能域以游离ER基因形式存在[21]。但这一还原酶特异性识别某些特定位置进行还原反应的原因尚不明确。
2.3 β-branch形成
trans-AT除能够通过甲基转移酶功能域催化聚酮化合物α位上发生取代反应而产生碳支链外,也存在β位酮基基团被转换成碳支链的现象,这两个不同位置的取代反应需要两种完全不同的机制。通过大量含有β-branch的聚酮化合物研究发现,β-branch由一组酶催化的与甲羟戊酸生物合成途径相似的β-酮基与丙二酰单元发生羧基缩合反应形成。这种有脱羧功能的酮基合成酶(KSQ)、羟甲基戊二酰辅酶A合成酶(HCS)、烯酰辅酶A脱水酶(ECH)和酰基载体蛋白(ACP)组成的负责β-branch形成的一组酶称作HCS卡壳(Hydroxy⁃methyleglutaryl-CoA synthase cassette)。通过酰基转移酶识别丙二酰辅酶A并将其装载到游离酰基载体蛋白上,通过KSQ催化一步脱羧反应,进行HCS催化缩合反应并在ECH作用下脱去一分子水,最终形成β位侧链。Haines等研究发现,ACP上的1个相对保守序列是β-branch形成的识别位点,ACP第3个螺旋结构对ACP和HCS形成起决定作用[22]。因此,通过ACP结构改造可在特定位置上添加β支链(见图2A)。
除通过HCS卡壳形成β-branch外,近年还在rhizoxin和iso-migrastatin生物合成途径中鉴定出一种全新的、与HCS不同的β-branch形成方式[23],最终形成δ-内酯和戊二酰亚胺类官能团。Hert⁃Weck等通过体内突变试验和分离鉴定生物合成途径利用中间体方法提出假设,即δ-内酯通过一个迈克尔加成反应将丙二酰单元与α,β不饱和硫脂结合形成。对预测的与β-branch形成相关的β-branching module(KS-B-ACP)作体内试验并在体外成功模拟侧链形成反应,最终确定侧链C-C键形成是通过一个共轭加成反应及δ-羟基攻击KS上的硫脂最终形成δ-内酯结构[24]。通过该蛋白晶体结构发现,C-C键形成和内酯化主要通过KS催化,然而首次发现的支链功能域(B domain)并未直接参与支链形成,所以B domain起辅助作用,引导KS完成β位侧链形成而非常规二碳单元延伸(见图2B)。Hertwech等通过体外试验证明戊二酰亚胺类官能团产生机理与之相同[25]。
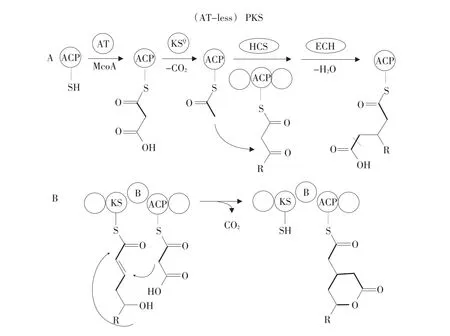
a-类似萜类化合物的β支链形成过程;b-乙烯β支链形成过程;KSQ-非延伸型脱羧酶;HCS-羟甲基戊二酰辅酶;A-合成酶;ECH-脱氢/脱羧酶;B-支链功能域a-Terpenoid-like b-branching; b-Vinylogous chain branching;KSQ-Non-elongating, decarboxylating ketosynthase; HCS-Hydroxymethylglutaryl-CoA synthase; ECH-Dehydratase/decarboxylase; B-Branching domain图2聚酮化合物β支链形成机制Fig. 2 Mechanisms of polyketide chain branching
2.4特殊功能的功能域
除上文描述的特殊功能域外,trans-AT型聚酮合酶基因簇中还存在多种结构特殊、功能新颖的功能域。例如Leinamycin生物合成聚酮合酶LnmJ包含两个与其他已知聚酮合酶功能域不同的特殊功能域,这两个功能域位于TE domain之前,推测其可能与Leinamycin上活性官能团的二硫键形成有关,但目前关于其功能的研究尚无报道[26]。此外,在Pederin、Onnamides和Bryostatins的trans-AT聚酮合酶基因簇中均包含不常见的PS功能域(Pyran synthase domain),此功能域最初被认为与脱水酶具有同源性,在该功能域所处的module上已存在1 个DH domain,因此被注释为PS domain,与常规DH domain加以区分。预测这个功能域可催化与常规DH domain完全相反的反应,即催化一分子乙醇与一分子不饱和硫脂之间的加成反应形成罕见的四氢呋喃环[12]。在含有类似呋喃环结构的on⁃namides和bryostatins的生物合成基因簇中也发现相似的PS domain,可侧面证明PS domain功能。bryo⁃ statins聚酮合酶基因簇上还存在两个不常见的OMT domain(O-methyltransferase domain)即氧甲基转移酶功能域,根据整个聚酮合酶基因簇和bryo⁃statins之间关系,推测OMT domain功能是在β-branch上面形成甲基酯[27]。同时在oxazolomycin 和thailandamides聚酮合酶基因簇中也发现与该OMT domain同源的domain,然而这三个结构相似的OMT domain在各自生物合成途径中却起到完全不同的功能,oxazolomycin中的OMT domain向α位的甲基中引入一分子氧而形成氧甲基[13],thailand⁃amides中的OMT domain则是在β位上的酮基上引入一分子甲基形成β位烯醇甲酯,再经额外的一步还原反应最终形成β位氧甲基基团[19]。2014年,在Burkholderia sp. FERM BP-3421基因组中发现1个与spliceostatins生物合成相关的基因簇,通过该基因簇注释,发现为1个trans-AT型聚酮合酶基因簇,该基因簇中除含有两个PS domain、4个非延伸功能的KS0、两个与β-branch相关的ECH domain和大量2个或者3个迭代的ACP外,基因簇内部还存在1个与三元氧环形成相关的FMO domain(Fla⁃vin-dependent monooxygenase),即黄素依赖性单加氧酶功能域。随后通过体内敲除试验确定FMO do⁃main功能并将其与化合物结构上的三元氧环形成关联[6]。除不常见的domain外,在trans-AT聚酮合酶基因簇中,如bacillaene生物合成基因簇中第三个module中的KR domain除能够催化常规β位酮基还原反应外,还催化α位上的酮基还原反应[28]。证明在trans-AT聚酮合酶基因簇中,普通domain也可重复作用或作用在远端位置。
3聚酮化合物筛选和结构改造的展望
传统化合物分离主要通过环境微生物分离纯化后,经多种培养基发酵和发酵液富集,采用随机分离和活性介导方式分离并对化合物作结构鉴定。这两种方法在分离前,无法对所分离化合物作精准预测,易导致化合物重复分离,同时微生物中的基因簇可能在某一特定培养基中处于沉默或表达较低水平,因此随机尝试培养基无法全面了解微生物次级代谢产生能力。随基因组测序技术发展,从基因组水平入手,以已知生物合成途径为基础对基因组中各个基因簇预测并通过不同分子生物学手段将目的基因簇异源表达,例如全长RecE介导的linear-linear同源重组系统[29]和同源重组效率较高的酵母TAR-cloning的利用[30]。针对性分离所需化合物,选取的异源表达宿主可降低发酵液复杂程度,便于化合物检测和分离。
以上特殊功能域研究,可更清晰认识聚酮化合物生物合成,结合已发表基因组序列,可从繁杂基因组数据中找到所需含有特殊功能域基因簇。在此基础上对多种基因簇修饰和改造,甚至打散重新组合达到自然进化目的,产生全新结构小分子药物。优化利用主要包括:①通过Ⅰ型聚酮合酶结构研究,已确定除MT外所有功能域活性位点[31],对活性功能域以突变失活方式改造基因簇,对碳链饱和程度人为修改产生非天然化合物。例如,通过对KR和DH功能域点突变失活后,分离得到非天然Candicidin类似物[32];②通过对AT功能域改造使聚酮合酶可识别不同前体从而改造化合物结构。例如,通过对erythromycin生物合成基因簇上的第6个AT功能域改造后,使聚酮合酶识别含有一个炔键侧链的丙二酸盐,并将其整合到erythromycin生物合成途径中,最终在红霉素结构中引入含炔键侧链[33]。在此基础上,通过体内突变试验将聚酮合酶上的AT功能域突变失活后,引入1个trans-AT并将氟乙酸盐衍生的氟丙二酰辅酶A整合到聚酮化合物的生物合成过程中,产生全新含氟短链聚酮化合物[34]。在药物化学研究中,氟原子具有较强生物活性,可在所需位置引入氟原子。2014年,Ye等研究trans-AT聚酮合酶中具有非甲基丙二酰活性的酰基转移酶KirCII和酰基载体蛋白蛋白结构并确定了内部关键位点[35]
近年来,trans-AT型聚酮合酶作为结构复杂且具有活性天然产物的重要来源被日益关注。由于trans-AT型聚酮合酶具有全新、多样性的催化方式,主要存在于特殊微生物中,与cis-AT聚酮合酶相比,trans-AT聚酮合酶研究仍处于起步阶段,其蛋白结构研究可解释各种酶或功能域的特殊功能,为改造和创造新的生物合成基因簇提供理论基础。
[参考文献]
[ 1 ] Staunton J, Weissman K J. Polyketide biosynthesis: A millennium review[J]. Nat Prod Rep, 2001, 18(4): 380-416.
[ 2 ] Smith S, Tsai S C. The type I fatty acid and polyketide synthases: A tale of two megasynthases[J]. Nat Prod Rep, 2007, 24(5): 1041-1072.
[ 3 ] Khosla C, Tang Y, Chen A Y, et al. Structure and mechanism of the 6-deoxyerythronolide B synthase[J]. Annu Rev Biochem, 2007, 76(1): 195-221.
[ 4 ] Partida-Martinez L P, Hertweck C. A gene cluster encoding rhi⁃zoxin biosynthesis in "Burkholderia rhizoxina", the bacterial en⁃dosymbiont of the fungus Rhizopus microsporus[J]. Chem Bio⁃chem, 2007, 8(1): 41-45.
[ 5 ] Lim S K, Ju J, Zazopoulos E, et al. Iso-Migrastatin, migrastatin, and dorrigocin production in Streptomyces platensis NRRL 18993 is governed by a single biosynthetic machinery featuring an acyl⁃transferase-less type I polyketide synthase[J]. J Biol Chem, 2009, 284(43): 29746-29756.
[ 6 ] Eustáquio A S, Janso J E, Ratnayake A S, et al. Spliceostatin hemiketal biosynthesis in Burkholderia spp. is catalyzed by an iron/α-ketoglutarate-dependent dioxygenase[J]. P Natl Acad Sci USA, 2014, 111(33): 3376-3385.
[ 7 ] Sherif I E, Amaro E T, Amro H, et al, Boronated tartrolon antibiot⁃ic produced by symbiotic cellulose-degrading bacteria in ship⁃worm gills[J]. P Natl Acad Sci USA, 2013, 110(4): 295-304.
[ 8 ] Cheng Y Q, Tang G L, Shen B. Type I polyketide synthase requir⁃ing a discrete acyltransferase for polyketide biosynthesis[J]. P Natl Acad Sci USA, 2003, 100(6): 3149-3154.
[ 9 ] Scotti C, Piatti M, Cuzzoni A, et al. A Bacillus subtilis large ORF coding for a polypeptide highly similar to polyketide synthases[J]. Gene, 1993, 130(1): 65-71.
[10] Paitan Y, Alon G, Orr E, et al. The first gene in the biosynthesis of the polyketide antibiotic TA of Myxococcus xanthus codes for a unique PKS module coupled to a peptide synthetase[J]. Journal of molecular biology, 1999, 286(2): 465-474.
[11] Huang G, Zhang L, Birch R G. A multifunctional polyketide-pep⁃tide synthetase essential for albicidin biosynthesis in Xanthomon⁃as albilineans[J]. Microbiology, 2001, 147(3): 631-642.
[12] Piel J. A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles[J]. P Natl Acad Sci USA, 2002, 99(22): 14002-14007.
[13] Zhao C, Ju J, Christenson S D, et al. Utilization of the methoxymal⁃onyl-acyl carrier protein biosynthesis locus for cloning the oxa⁃zolomycin biosynthetic gene cluster from Streptomyces albus JA3453[J]. Journal of Bacteriology, 2006, 188(11): 4142-4147.
[14] Pulsawat N, Kitani S, Nihira T. Characterization of biosynthetic gene cluster for the production of virginiamycin M, a strepto⁃gramin type A antibiotic, in Streptomyces virginiae[J]. Gene, 2007, 393(1-2): 31-42.
[15] Mochizuki S, Hiratsu K, Suwa M, et al. The large linear plasmid pSLA2-L of Streptomyces rochei has an unusually condensed gene organization for secondary metabolism[J]. Mol Microbiol, 2003, 48 (6): 1501-1510.
[16] Weber T, Laiple K J, Pross E K, et al. Molecular analysis of the kirromycin biosynthetic gene cluster revealed β-alanine as pre⁃cursor of the pyridone moiety[J]. Chemistry & Biology, 2008, 15 (2): 175-188.
[17] Piel J, Hui D Q, Fusetani N, et al. Targeting modular polyketide synthases with iteratively acting acyltransferases from metage⁃nomes of uncultured bacterial consortia[J]. Environ Microbiol, 2004, 6(9): 921-927.
[18] Jenke-Kodama H, Boerner T, Dittmann E. Natural biocombinator⁃ics in the polyketide synthase genes of the actinobacterium Strep⁃tomyces avermitilis[J]. PLoS Comput Biol, 2006, 2(10): 1210-1218.
[19] Nguyen T, Ishida K, Jenke-Kodama H, et al. Exploiting the mosa⁃ic structure of trans-acyltransferase polyketide synthases for natu⁃ral product discovery and pathway dissection[J]. Nature Biotech⁃nology, 2008, 26(2): 225-233.
[20] Hill A M. The biosynthesis, molecular genetics and enzymology of the polyketide-derived metabolites[J]. Nat Prod Rep, 2006, 23(2): 256-320.
[21] Bumpus S B, Magarvey N A, Kelleher N L, et al. Polyunsaturated fatty-acid-like trans-enoyl reductases utilized in polyketide bio⁃synthesis[J]. J Am Chem Soc, 2008, 130(35): 11614-11616.
[22] Haines A S, Dong X, Song Z, et al. A conserved motif flags acyl carrier proteins for beta-branching in polyketide synthesis[J]. Na⁃ture Chemical Biology, 2013, 9(11): 685-692.
[23] Kusebauch B, Busch B, Scherlach K, et al. Polyketide-chain branching by an enzymatic Michael addition[J]. Angew Chem Int Ed Engl, 2009, 48(27): 5001-5004.
[24] Bretschneider T, Heim J B, Heine D, et al. Vinylogous chain branching catalysed by a dedicated polyketide synthase module [J]. Nature, 2014, 502(7469): 124-128.
[25] Heine D, Bretschneider T, Sundaram S, et al. Enzymatic polyketide chain branching to give substituted lactone, lactam, and glutarim⁃ide heterocycles[J]. Angew Chem Int Ed Engl, 2014, 53(43): 11645-11649.
[26] Piel J. Biosynthesis of polyketides by trans-AT polyketide syn⁃thases[J]. Nat Prod Rep, 2010, 27(7): 996-1047.
[27] Sudek S, Lopanik N B, Waggoner L E, et al. Identification of the putative bryostatin polyketide synthase gene cluster from "Candi⁃datus Endobugula sertula," the uncultivated microbial symbiont of the marine bryozoan Bugula neritina[J]. Journal of Natural Prod⁃ucts, 2007, 70(1): 67-74.
[28] Calderone C T, Bumpus S B, Kelleher N L, et al. A ketoreductase domain in the PksJ protein of the bacillaene assembly line carries out both alpha- and beta-ketone reduction during chain growth [J]. P Natl Acad Sci USA, 2008, 105(35): 12809-12814.
[29] Fu J, Bian X, Hu S, et al. Full-length RecE enhances linear-lin⁃ear homologous recombination and facilitates direct cloning for bioprospecting[J]. Nature Biotechnology, 2012, 30(5): 440-446.
[30] Yamanaka K, Reynolds K A, Kersten R D, et al. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A[J]. P Natl Acad Sci USA, 2014, 111(5): 1957-1962.
[31] Keatinge-Clay A T. The structures of type I polyketide synthases [J]. Nat Prod Rep, 2012, 29(10): 1050-1073.
[32] Zhou Y, Li J, Zhu J, et al. Incomplete β-Ketone processing as a mechanism for polyene structural variation in the FR-008/candi⁃cidin complex[J]. Chemistry & Biology, 2008, 15(6): 629-638.
[33] Sundermann U, Bravo-Rodriguez K, Klopries S, et al. Enzyme-di⁃rected mutasynthesis: A combined experimental and theoretical approach to substrate recognition of a polyketide synthase[J]. ACS Chem Biol, 2013, 8(2): 443-450.
[34] Walker M C, Thuronyi B W, Charkoudian L K, et al. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways[J]. Science, 2013, 341(6150): 1089-1094.
[35] Ye Z, Musiol E M, Weber T, et al. Reprogramming acyl carrier protein interactions of an Acyl-CoA promiscuous trans-acyltrans⁃ferase[J]. Chemistry & Biology, 2014, 21(5): 636-646.
Advances in Cis-AT and Trans-AT polyketide synthases and their special domains
ZHANG Bo, XIANG Wensheng(School of Life Sciences, Northeast Agricultural University, Harbin 150030, China)
Abstract:Due to the diversity and specificity of polyketide synthase (PKS), the polyketide natural products generated by PKSs were structurally and bioactively diversified. Recently, the trans-AT polyketide synthase was discovered, characterized and investigated as the subfamily of TypIe I PKSs. In this review, the difference between cis-AT PKS and trans-AT PKS as well as the research advance of special domains in trans-AT PKS were summarized and discussed. In addition, the methods could be use to modify the gene clusters to generate the novel structure compounds were also analyzed and providing the new target for natural product modification.
Key words:trans-AT polyketide synthase; special domains; structural improvement
*通讯作者:向文胜,教授,博士生导师,研究方向为微生物农药。E-mail:xiangwensheng@neau.edu.cn
作者简介:张博(1986-),男,博士研究生,研究方向为微生物天然产物生物合成。E-mail:bozhang95@yahoo.com
收稿日期:2015-07-29
中图分类号:Q78
文献标志码:A
文章编号:1005-9369(2016)01-0087-06
