印度对医疗器械市场准入的要求
2016-03-16秦韶燕崔涛殷海松
【作 者】秦韶燕,崔涛,殷海松
1 蓝莫德(天津)科学仪器有限公司,天津市,300384
2 天津市医疗器械技术审评中心,天津市,300191
3 天津现代职业技术学院生物工程学院,天津市,300350
印度对医疗器械市场准入的要求
【作 者】秦韶燕1,崔涛2,殷海松3
1 蓝莫德(天津)科学仪器有限公司,天津市,300384
2 天津市医疗器械技术审评中心,天津市,300191
3 天津现代职业技术学院生物工程学院,天津市,300350
该文简要介绍了印度医疗器械市场准入的注册程序、应提交的注册文件以及上市后的监管要求。依据印度医疗器械法和医疗器械相关法规,就其对进口医疗器械的注册管理制度进行了初步探讨。旨在为国外医疗器械进入印度市场作为参考。
医疗器械;市场准入;印度
0 引言
在世界上,印度是人口仅次于中国的发展中国家,因此,医疗器械市场潜力巨大。而且印度医疗保障制度与中国不同,印度坚持两手抓,一方面扶持政府医院的稳定运转,另一方面又积极鼓励私立医院健康发展。因此,在印度医院的现状是政府医院床位比较短缺,相反很多的私立医院医疗设备非常齐全,甚至医疗条件并不亚于欧洲的医院[1]。
随着印度市场医疗产业的发展,市场对医疗器械的需求也在不断的提高。印度医疗器械产业存在巨大的市场空间,市场前景非常广阔。中国作为第三大医疗器械产品的供货商,能够以较低的生产成本生产具有价格优势的医疗器械,并有规模化生产的出口优势,占据印度市场潜力巨大[2]。
然而,印度对医疗器械管理比较混乱,而且规章繁多,外国人很难弄清楚其中的头绪。而且与印度政府官员沟通也是一大难题。据印度有关媒体报道,截至2006年为止,印度政府从未颁布过一部完整的关于医疗器械的政策法规。这让国外厂商感到无所适从。由于对印出口医疗器械不仅手续繁琐复杂,而且时间很长,因此,向印度这样的国家出口医疗器械产品不是一件容易的事情[3]。
本文通过对印度医疗器械法规、规章、条文的学习和理解,结合实际工作经验,总结了印度医疗器械市场准入的要求,以供相关人员参考、交流和探讨。
1 医疗器械的监管[4]
中央药品标准控制机构(Central Drugs Standard Control Organization,CDSCO),是印度卫生与福利部(Ministry of Health and Family Welfare)下属的监管医疗器械进口、生产和销售的重要部门。中央药品标准控制机构曾经是管理药品、诊断试剂和化妆品的标准、法规、生产和进口的部门。现在,CDSCO的职能是制定药品、疫苗、血液制品和注射液的标准、法规。同时该机构增加了监管医疗器械的职能,为了加强对医疗器械的监督管理。
在多数国家,有专门机构对医疗器械进行法规监管,而且在多数国家的分类管理中,医疗器械并不属于药品。但是印度没有专门的医疗器械法规,而是按照《药品和化妆品法》来对医疗器械进行管理。在印度《药品化妆品法》是对医疗器械监督管理的基本法。
2 医疗器械的定义和分类
2.1 医疗器械的定义
在印度,医疗器械没有单独的定义,与中国医疗器械的分类管理[5]不同,在印度医疗器械是作为药品进行管理。在印度的《药品和化妆品法》[6]中明确写明,药品包括下列四项,而第四项为医疗器械:
(1) 人类或动物内服或外用,用于人类和动物疾病的诊断、治疗、减缓或预防的物质,包括用于人体驱虫的物质。
(2) 预期影响人体结构或功能,用于破坏导致人类和动物疾病的物质(不包括食品)。
(3) 作为药物的组成成分,例如:空胶囊。
(4) 用于人体或者动物内用或者外用,以诊断、治疗、预防或缓解疾病和异常为目的的器械。
2.2 医疗器械的分类
在印度,《药品和化妆品法》和相关的药品监管法规没有按风险水平对医疗器械进行分类。所以,很多医疗器械的监管情况都与印度管理药品的法规密不可分。
3 医疗器械上市前许可
3.1 确认医疗器械是否需要注册[7]
在印度,不是所有的医疗器械都需要注册,只有公告的医疗器械才需要注册,主要包括表1中的14类。
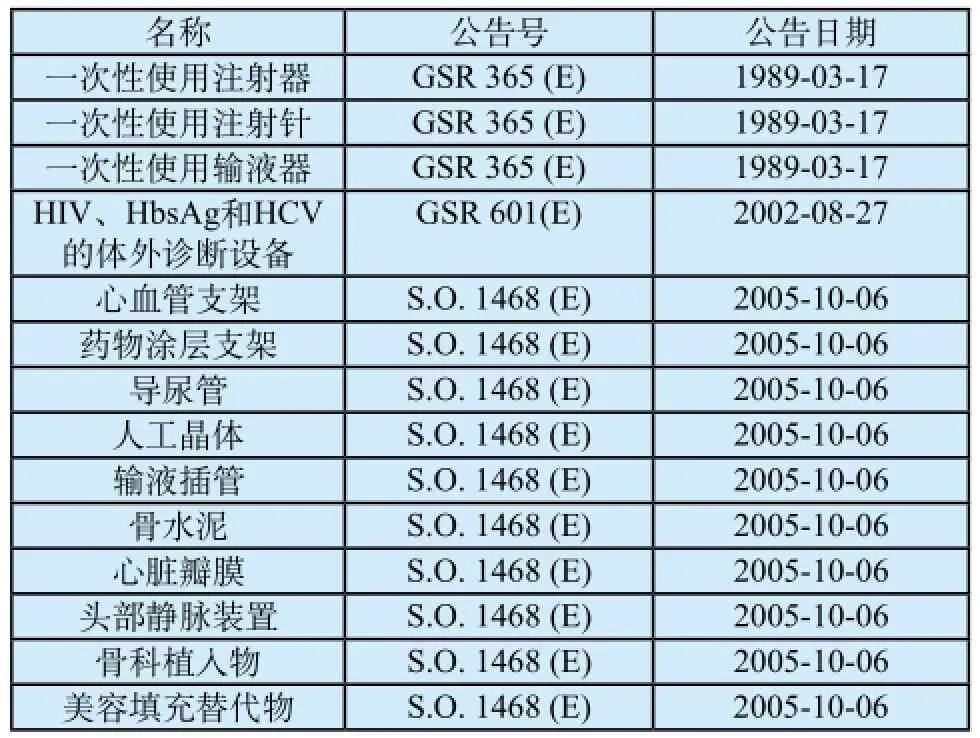
表1 公告医疗器械列表Tab.1 The list of notified medical device
未公告的医疗器械进入印度不需要进行注册。但是,需要强调的是,下面这些器械在印度作为药品管理,因此,这些产品进入印度市场也需要注册:①与血型检测相关试剂(Blood Grouping Sera);②绷带、缝合线以及吻合器类产品(Ligatures, Sutures, Staples);③宫内节育器(Intra Uterine Devices (Cu-T));④避孕套(Condoms);⑤输卵管环(Tubal Rings);⑥敷料(Surgical Dressing);⑦婴儿脐带结扎产品(Umbilical Tapes);⑧血液/血液成分储存袋(Blood / Blood Component Bags)。
3.2 医疗器械在印度注册的条件[8]
如果医疗器械要在印度申请注册,首先该产品必须已经通过以下至少一种认证,申请资料中应包括相应的认证证书:①美国FDA认证;②澳大利亚TGA认证;③日本JAPL认证;④加拿大HC认证;⑤欧盟的CE认证。
另外,如果预申请的医疗器械未在原产地注册,但是已有以上五种认证中的任意一种,也可以在印度申请进口注册。
同时,需要注意的是,也并非所有未通过任意一种以上认证的产品就完全不可以在印度注册,该产品需要在印度本地做临床试验,通过临床试验证明了产品的安全性和有效性后,可以申请产品注册。但是,在印度做临床试验是一件非常耗费时间的事情。
3.3 进口医疗器械注册的步骤
医疗器械要在印度上市,需要申请医疗器械注册证,注册证书有效期为三年,需要在证书有效期九个月前提交重新注册申请。医疗器械注册主要分为四步:
第一步:支付费用。制造商的企业注册需要支付1 500美金。每个产品的注册需要1 000美元。
第二步:准备资料。印度进口医疗器械注册主要参考文件为《印度医疗器械注册指南》[9],以下简称指南。该指南中详细介绍了医疗器械注册需要准备的文件,以及文件中需要包括的具体内容。其中企业主文档(Plant/ Site Master File)和产品主文档(Device Master File)为其中涵盖内容最多也是最重要的两份文件。需要准备文件如下:
(1) 附信(Covering Letter)。随提交文件一起提交的说明信。这封说明信非常重要,信中应详细说明产品的注册是首次注册还是重新注册,还是扩增产品注册。
(2) 申请表(Form 40)。申请表中包括制造商信息,申请产品信息,以及需要交纳费用等信息。
(3) 原始收据(Receipt in original)。缴纳注册相关费用的原始收据。
(4) 授权信(Power of Attorney)。制造商授权印度代理商的一份证明性文件,内容包括双方的公司名称地址,要注册产品的名称型号等信息,模板可参考指南的附录III。应注意,授权信需要在制造商所在地国家的印度使馆进行认证。
(5) 自由销售证明书(Free Sale Certificate)。原产地的自由销售证明书或在美国、加拿大、日本、澳大利亚和欧盟的自由销售证明书(如适用)。原产地的自由销售证明书应在制造商所在地国家的印度使馆进行认证。
(6) 依据ISO13485颁发的体系证书(ISO 13485Certificate)。
(7) CE证书(CE Full Quality Assurance Certificate)。应在制造商所在地国家的印度使馆进行认证。
但也有人以为,这是洋人预先做了什么手脚在里边,不甚相信听到的说话声音,是从几里外的正丰街或十六铺传过来的,他们计划拆穿外国佬的西洋镜。于是呼朋唤友,或邻居相邀,约好通话时第一句讲什么,第二句讲什么,诸如问答姓名、住址,或是去哪里等等。
(8) CE设计确认证书(CE Design Certificate),(如适用)。只要在欧盟分类为第三类的产品才有CE设计确认证书。也应在制造商所在地国家的印度使馆进行认证。
(9) 符合性声明(Declaration of Conformity)。
(10) 审核报告(Inspection/Audit Report)。
(11) 企业主文档(Plant/ Site Master File)。
在指南附录V中对企业主文档的要求做了详细的说明。从企业概况,生产产品的介绍到企业机构与人员、厂房和设施、设备、生产、质量控制、储存、文件、抱怨和纠正预防措施等各个方面的要求,全面详细地介绍体系文件。虽然文件与ISO 13485非常相似,但是要按照附录V的要求整理一份企业主文档,是一件费时费力的事情。
同时,在《印度医疗器械进口注册问题汇总》[10]中47条说到,按照ISO 13485制定的《质量手册》,在医疗器械注册中可提交质量手册来替代按照指南附录V准备的企业主文档。
(12) 产品主文档(Device Master File)。
按照指南的附录VI编写产品主文档。包括内容基本与CE认证技术文档一致,但是要按照附录VI的要求编写,主要内容有:
① 产品综述:简要介绍产品预期用途;结构;有效期;是否无菌;价格;在其他国家的注册情况;不良事件以及整改措施等;
② 详细的产品描述以及规格介绍:产品预期用途;禁忌症;材质;适用人群;结构说明;附件的说明;主要功能部件的说明;规格型号的详细区别及描述;以及对比产品的情况说明。
③ 包装标签:主要包括产品的宣传册,操作手册,以及产品的包装(应包括产品所有附件的包装以及标签)。按照印度《药品和化妆品法》,注册时要提供一整套产品的包装原件(应包括产品所有附件的包装以及标签)。
④ 设计和生产信息:设计资料包括产品生产流程图和产品设计确认资料。生产信息包括产品生产概况;生产环境;生产设备;产品组装;半成品、成品检验;产品的包装及储存信息。如果产品的生产在不同的生产地址,应对不同生产地址进行有效的区别。
⑤ 适用基本标准列表:该产品适用哪些标准,以及相关的检测报告或符合性说明。
⑥ 风险分析报告。
⑦ 产品确认和验证报告:包括工艺验证报告;生物安全性试验;无菌验证报告;软件验证确认报告;动物试验;加速老化试验;临床资料;上市后的检测数据。
第三步:提交产品注册申请到CDSCO。
第四步:获证。如果提交资料齐全,CDSCO会在9个月之内颁发医疗器械注册证书。
4 医疗器械上市后监管[1]
医疗器械在印度属于药品,所以医疗器械上市后的监管也按药品的监督管理来进行。医疗器械上市之后,一旦出现不良事件,制造商必须向CDSCO报告。当医疗器械被强制撤出其他国家市场,或是取消合法许可时,也必须撤出印度市场,这种撤出是暂时性的,然后CDSCO会进一步讨论合理的管理活动。
制造商在申请注册时即被要求进行上市后监督管理,包括产品分销记录程序、投诉处理、不良事件报告和产品召回程序等。
5 总结
综上所述,国外医疗器械产品要进入印度市场,首先,需要确认产品是否需要注册;其次,判断该产品是否符合注册的要求;最后,按照《印度医疗器械注册指南》的要求,准备详细的注册资料。
以上为结合实际工作经验以及查看印度相关法规后总结所得,希望能为更多的医疗器械产品进入印度市场提供帮助。
[1] 潘尔顿, 宓现强. 印度医疗器械监管概述.中国医疗器械信息[J]. 2009, 15(4), 59-62.
[2] 陈蓉. 中国医疗器械出口印度市场的问题及对策[J]. 中外企业家, 2010(Z4):, 32-35.
[3] 徐镜. 外商眼中的印度医疗器械市场: 机会多多 赚钱不易[N].中国医药报, 2008-07-08(A08).
[4] 夏芸, 甄辉. 印度对进口医疗器械的要求[J]. 中国医疗器械信息, 2007, 13(10): 43-45.
[5] 崔怡, 崔亮, 赵京霞, 等. 医疗器械的分类管理[J]. 医疗卫生装备, 2013, (09): 125-126.
[6] The drugs and cosmetics act and rules[EB/OL]. [2005-06-30]. http://cdsco.nic.in/forms/contentpage1.aspx?lid=1888.
[7] The list of notified medical device[EB/OL]. [2012-04-20]. http:// cdsco.nic.in/forms/list.aspx?lid=1580&Id=1.
[8] Clarification on guidelines for import & manufacture of medical device[EB/OL]. [2007-9-5].http://cdsco.nic.in/Forms/list.aspx lid=1863.
[9] Guidance document on common submission format for registration/ re-registration of notified medical devices in India[EB/OL]. [2013-01-01].http://cdsco.nic.in/forms/list.aspx?lid=1674&Id=1.
[10] Frequently asked questions on registration and import of medical devices in India[EB/OL]. [2013-02-21].http://cdsco.nic.in/forms/ list.aspx?lid=1580&Id=1.
The Requirements of Medical Device Market Access in lndia
【 Writers 】QIN Shaoyan1, CUI Tao2, YIN Haisong3
1 Lanmode (Tianjin) Scientific Instrument Co. Ltd., Tianjin, 300384
2 Tianjin Medical Instrument Technical Evaluation Center, Tianjin, 300191
3 School of Bioengineering, Tianjin Modern Vocational Technology College, Tianjin, 300350
medical device, market access, India
F203
A
10.3969/j.issn.1671-7104.2016.01.018
1671-7104(2016)01-0061-03
2015-10-26
秦韶燕,E-mail: qinshaoyan521@163.com
【 Abstract 】This paper introduces the premarket registration procedures and the post market regulatory requirements in India. According to Indian medical device act and related medical regulations on medical device, this is a preliminary discussion on the registration management system to provide referance for foreign medical device to enter India market.
