碳掺杂WO3电子结构的第一性原理研究
2016-03-01许英韩丹
许英 韩丹

摘 要 采用第一性原理计算方法详细研究了碳原子掺入WO3后体系的能带结构、态密度、导带边和价带边的位置的变化.考虑了碳原子处在间隙位置和碳原子替换氧原子两种情况.计算结果表明,碳原子替换氧原子时,体系是自旋极化的,在带隙中产生非常明显的C-2p杂质带. 自旋向上部分带隙减小了0.92 eV, 自旋向下部分带隙减小了0.08 eV. 而碳处在间隙位置时,体系是非自旋极化的,碳原子倾向于与一个氧原子成键,带隙减小了0.2 eV,有利于可见光的吸收. 形成能计算表明,碳原子处在间隙位置掺杂更稳定.
关键词 第一性原理计算;掺杂;形成能;光催化活性
中图分类号 O4715 文献标识码 A 文章编号 1000-2537(2015)04-0057-06
Abstract The properties of carbon doped WO3 are calculated by first principles calculations. The electronic structure, the valence band maximum and conduction band minimum are studied. Both carbon substituting oxygen atom or staying in the interstitial site are considered. The calculation results show that the system is spin polarized with carbon substituting oxygen atom and an impurity C-2p band emerges in the middle of the band gap. The band gap decreases by 0.92 eV for spin-up part and 0.08 eV for spin-down part. When the impurity carbon atom stays in the interstitial site, the system is unpolarized and the carbon atom prefers to bond one oxygen atom. The band gap decreases by 0.2 eV, which is beneficial for absorption of visible light. Formation energy calculation shows that the carbon atom prefers to site in the interstitial site rather than the substituting oxygen sites.
Key words first-principles calculation; doping; formation energy; photocatalytic activities
1972年,日本学者Fujishima和Honda在Nature杂志上发表了TiO2电极光分解水的文章,标志着光催化时代的开始[1].光催化剂是光催化过程的关键部分.目前在光催化研究中所使用的光催化剂大都是半导体.半导体光催化剂必须具有合适的禁带宽度、导带和价带电位. TiO2因其化学性质稳定、抗光腐蚀能力强、难溶、无毒、低成本,是研究中使用最广泛的光催化材料.但是,TiO2的禁带宽度达3.2eV,光吸收仅限于紫外光区,只能响应占太阳光谱约5%左右的紫外光,太阳能利用率很低.再加上光生电子空穴对的复合率高,太阳能利用效率仅在1%左右.
因此,要提高太阳能利用效率,就必须扩大催化剂响应太阳光波长的范围,即减小半导体催化剂的带隙至2 eV左右. 这其中, WO3吸引了人们的注意.WO3是一种n型催化剂[2],具有较好的光敏性,良好的电子输运特征以及光腐蚀稳定性.并且相对于其他的光催化剂,例如TiO2,WO3,具有较小的带隙,2.8 eV, 适合吸收可见光,是一种非常有潜力的半导体光催化材料.将WO3应用在半导体催化剂领域,有两点亟待改进:一是WO3的带隙还是过大,不能实现对太阳光的有效吸收;二是实验结果显示块体WO3的导带边比氢的氧化还原势低0.4 eV(另有实验结果[3-4]低0.31 eV).因此对其进行能带调控的目的一是减小其带隙,二是抬高导带边,增加催化活性.实验发现Mg掺杂导致导带边变向上移动[5],Mo掺杂的WO3纳米线使带隙减小0.43 eV, 增加可见光的利用率[6].一些过渡金属,例如Ni掺杂产生了最大氢气[7]. 掺入其他金属如Ti, Zn, Dy, Te, Ta, V, Cu, Ag和Ce, 也被报道提高了WO3的光催化性质[8-12].
相对于TiO2, 目前利用掺杂对WO3进行能带调控的理论研究还非常少.文献[13]中,Huda等人采用PAW-LDA计算了N掺杂后WO3的电子结构变化.发现N掺杂后带隙的减小主要源于带隙中杂质态的形成,在文献[14] 中,Wang等人采用LCAO方法应用B3LYP杂化泛函,计算了Cr, Mo, Ti, Zr和Hf元素对W原子进行替换,非金属元素S对氧原子的替换,以及Hf+S、Hf+2H共掺杂对能带结构的影响.结果发现Hf+2F原子共掺杂为最佳,能够使价带边和导带边分别向上移动0.55 eV和0.17 eV,带隙减小0.38 eV.非金属元素N、C的掺杂都被证实能改变带隙[15-16]. Sun 等人通过喷雾热分解法(spray pyrolysis)方法合成C掺杂的WO3[17],发现WO3薄膜的带隙减小,与未掺杂相比,光电流密度提高了50%,增强了对可见光的吸收.WO3是一种非常有潜力的光催化材料,但是这方面的理论研究非常有限.对于非金属元素碳掺杂的理论研究还未见报道.而碳元素掺杂后,结构如何变化,如何影响WO3体系的能带结构,碳原子处于间隙位置还是替换氧原子的位置更稳定,是本文关心的问题. 采用第一性原理计算的方法,研究了碳原子掺入后WO3的能带结构、态密度、不同位置的形成能、价带边及导带边的位置变化及禁带宽度的大小.并基于这些性质讨论掺杂后WO3的光催化特性.
1 计算方法
采用基于密度泛函理论(density functional theory)的VASP[18-19]软件包进行计算. 电子与电子交换关联势采用局域密度近似(LDA),离子-电子间相互作用采用平面波赝势方法(PAW);平面波截断能设置为450 eV,能量收敛精度为1.0×10-6 eV,原子间相互作用力收敛至0.01 eV. 布里渊区积分计算采用Monkorst-Park 高对称特殊k点方法. 在结构优化过程中采用2×2×2 的k网格点,态密度积分计算则采用8×8×8的k网格点.考虑了掺杂后的碳原子替换氧原子位置和碳原子处在间隙位置的两种情况.
2 结果与讨论
2.1 WO3块体的能带结构和态密度
WO3块体是单斜结构,其原胞由8个W原子和24个氧原子组成.WO3块体可视作一种有缺陷的锐钛矿结构(类似于ABO3的形式),其中A原子被移除,B原子被W原子替代,如图1所示.WO3块体也可视作由W-O-W构成的链状结构. 该结构存在较大的空隙,有利于掺杂.晶格结构优化完成后得到WO3块体的晶格常数分别是a=7.380 ,b=7.419 ,c=7.640 .这些数值与实验条件下所得WO3块体的晶格常数(a=7.306 , b=7.540 ,c=7.692 )较为符合[20],与其他理论计算结果一致[21,13],说明计算精度可靠.在后续计算中固定该晶格常数.
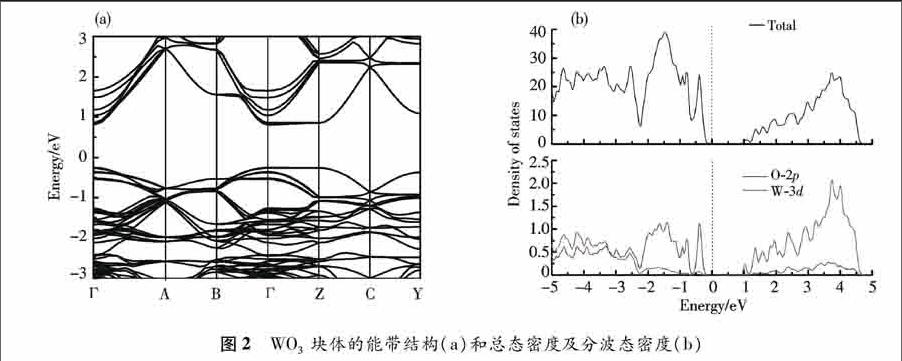
首先计算WO3块体能带结构和态密度. 从图2(a)可知WO3是直接带隙半导体,导带底和价带顶均位于布里渊区的Γ点处.禁带宽度为1.12 eV,该值远小于文献[10]报道的实验值2.70 eV.这是由于局域密度近似(LDA)的局限性,但不影响对于掺杂前后WO3体系能带结构变化规律的分析.图2(a)显示在Γ-Z, Γ-B方向能带色散很小,较为平坦,说明在这些方向,空穴的有效质量很大.而在其他方向,存在色散很大的带.图2(b)给出了WO3块体的总态密度和分波态密度.在费米面附近,价带的总态密度有个较尖锐的峰,对应费米能级附近较平滑的能带.O-2p带主要分布在费米能级以下至-2.2 eV能量范围.在-2.2 eV以下,可以观察到 O-2p带与W-5d带的较强杂化.W-5d带在费米能级1 eV以上有较大的峰.WO3块体的价带顶主要是由O-2p轨道贡献,导带底主要由W-5d轨道贡献.
2.2 碳原子替换氧原子后体系的能带结构和态密度
接下来计算碳原子在WO3原胞内取代氧原子后的电子结构.一个碳原子取代WO3原胞中的一个氧原子,对应碳原子的掺杂浓度为4%.碳原子替换氧原子后,C—W键长较原来C—O键长增大约1%,这是由于碳原子的半径略大于氧原子.由于存在未配对的电子,碳原子取代氧原子后,体系变成自旋极化.态密度如图3所示.因为碳原子比氧原子多两个价电子,费米能级落在导带边上.C-2p态产生了较大的自旋劈裂, 碳原子上产生了0.64 μB的磁矩.最近,已有不少文献报道了这种半导体中非金属元素如氮、碳掺杂带来的磁性[22-24].自旋向上部分在费米能级下形成一个尖锐的杂质峰,这一杂质峰主要由C-2p构成.并且C-2p, O-2p, W-5d有明显的杂化.价带边也主要由C-2p带贡献.从能带图(图3a)可以清楚地看到C-2p态在带隙中形成了杂质带.碳替换以后,体系的价带顶和导带底仍然在Γ点.带隙较之未掺杂前明显减小.自旋向上的部分带隙减小到只有0.3 eV,自旋向下的部分带隙减小到1.04 eV.
2.3 碳原子处在间隙位置时体系的能带结构和态密度
碳原子在间隙位置掺杂的结构如图4(a)所示.我们发现弛豫以后,碳原子周围的环境发生了较大的畸变,某一氧原子向内弛豫,与钨原子的成键键长增加为原来的8%,变成与碳原子成键,键长为1.293 .因此碳原子在间隙位置的掺杂可视作一个CO分子取代了一个氧原子.与碳原子替换氧原子掺杂不同,碳原子在间隙位置掺杂,体系是非自旋极化的.能带结构和态密度图如图4(b)和(c)所示.发现碳原子在间隙位置掺杂后,WO3未产生自旋极化现象.由于碳原子的掺入带来了更多电子,费米能级落在导带边上,体系表现出典型的n-型掺杂的特征.碳原子的2p轨道与氧原子的2p轨道有显著的杂化. 与未掺杂相比,导带顶主要仍由W-5d 贡献,而价带顶主要是C-2p和O-2p贡献.碳原子在间隙位置掺杂后,从能带图可看出,较掺杂前,能带的结构没有明显的改变,只是禁带宽度也较未掺杂前减小,为0.92 eV.相对于未掺杂前,减小了0.2 eV,这与实验观察碳掺杂后光吸收谱的红移是一致的[17].
2.4 讨论
为了比较碳原子在间隙位置和在替换位置的稳定性,计算了碳原子在两种不同位置时的形成能.形成能定义为[25]
从表1的结果来看, Ci在碳丰富的情况下稳定,而CO则在各化学条件下都不稳定,能量高于Ci.因此,C掺入WO3后,倾向于进入间隙位置.这与实验的结果一致[17].
半导体材料的光催化活性与下列因素密切相关:能带边的位置、被吸附物质的还原势及光生载流子的迁移能力[18].因此掺杂后价带顶和导带底的位置非常重要.为了进一步了解碳原子掺杂后体系的光催化活性,本文根据经验公式(2)和(3)计算了上述原子掺杂后的WO3体系的能带边位置[28-29].
式中的ECB代表导带边的势能值,X代表半导体的电负性大小,即体系内所有原子的亲和能和第一电离能平均数的几何平均值,Ee代表自由电子的能量约为4.5 eV,Eg是半导体体系禁带宽度的大小,掺杂后体系的Eg可以通过剪刀公式(Eg=E′g+ΔE)进行修正,其中E′g表示理论计算所得的掺杂体系的禁带宽度,ΔE为未掺杂体系实验所得的禁带宽度与理论计算得到的禁带宽度的差值.(3)式中的 EVB代表价带边的势能值,ECB由(2)式计算得到.计算结果发现,C替换O后,体系的价带边升高了0.34 eV,导带边降低了0.484 eV,而C处在间隙位置时,体系的导带边升高了0.111,价带边降低了0.089 eV. 在光催化剂催化水分解过程中,WO3块体价带边所对应的能量值比产生氧气所需的势能值要低很多,有利于产生氧气,但导带边所对应的能量值仍比产生氢气所需要的势能值低,不利于产生氢气.为了充分利用可见光,需减小WO3块体的禁带宽度.由此可知,价带边的升高说明掺杂后体系的氧化能力增强,导带边下降表明掺杂后体系的还原能力减弱.因此碳原子处在间隙位置时,能使带隙减小,价带边上升,但是不足之处是使导带边也略为下降.
3 结论
采用第一性原理计算方法分别详细研究了碳原子掺入WO3后,体系的能带结构、态密度、导带边和价带边的位置变化.考虑了碳原子处在间隙位置和碳原子替换氧原子两种情况.计算结果表明,碳原子替换氧原子时,体系是自旋极化的,在带隙中产生非常明显的C-2p杂质带.体系的价带边升高了0.34 eV,导带边降低了0.484 eV, 自旋向上部分带隙减小了0.92 eV, 自旋向下部分带隙减小了0.08 eV. 而碳处在替换位置时,体系是非自旋极化的,碳原子倾向于与一个氧原子成键,体系的导带边升高了0.111,价带边降低了0089 eV,带隙减小了0.2 eV,有利于可见光的吸收,与实验结果一致. 形成能分析表明,碳处在间隙位置掺杂更稳定. 计算结果对进一步提高WO3催化性能有理论上的指导意义.
参考文献:
[1] FUJISHIMA A, HOMDA K. Electrochemical photolysis of water at a semiconductor electrode [J]. Nature, 1972,238(5358):37-38.
[2] MILLER E L, MARSEN B, COLE B, et al. Electrochem, low-temperature reactively sputtered tungsten oxide films for solar-powered water splitting applications [J]. Solid-State Lett, 2006,9(7):248-250.
[3] NOZIK, ANNU A. Photoelectrochemistry: applications to solar energy conversion [J]. Ann Rev Phys Chem, 1978,29(1):189-222.
[4] WEINHARDT L, BLUM M, HESKE C, COLE B, et al. Electronic surface level positions of WO3, thin films for photoelectrochemical hydrogen production [J]. J Phys Chem C, 2008,112(8):3078-3082.
[5] HWNAG D W, KIM J, PARK T J, et al. Mg-doped WO3 as a novel photocatalyst for visible light-induced water splitting [J]. Catal Lett, 2002,80(1-2):53-57.
[6] ZHOU L, ZHU J, YU M, et al. MoxW1-xO3·0.33 H2O solid solutions with tunable band gaps [J]. J Phys Chem C, 2010,114(49):20947-20954.
[7] HAMEED A, GONDAL M A, YAMANI Z H. Effect of transition metal doping on photocatalytic activity of WO3 for water splitting under laser illumination: role of 3d-orbitals [J]. Catal Commun, 2004,5(11):715-719.
[8] RADECKA M, SOBAS P, WIERZBICKA M, et al. Photoelectrochemical properties of undoped and Ti-doped WO3[J]. Phys B, 2005,364(1-4):85-92.
[9] CHENG X F, LENG W H, LIU D P, et al. Enhanced photoelectrocatalytic performance of Zn-doped WO3 photocatalysts for nitrite ions degradation under visible light[J]. Chemosphere, 2007,68(10):1976-1984.
[10] LIU H, PENG T, KE D, et al. Preparation and photocatalytic activity of dysprosium doped tungsten trioxide nanoparticles [J]. Mater Chem Phys, 2007,104(2-3):377-383.
[11] YANG B, LUCA V. Enhanced long-wavelength transient photoresponsiveness of WO3 induced by tellurium doping[J]. Chem Commun, 2008,(37):4454-4456.
[12] ENESCA A, DUTA A. Schoonman, influence of tantalum dopant ions (Ta5+) on the efficiency of the tungsten trioxide photoelectrode[J]. J Phys Stat Solid A, 2008,205(8):2038-2041.
[13] HUDA M N, YAN Y, MOON C Y, et al. Density-functional theory study of the effects of atomic impurity on the band edges of monoclinic WO3 [J]. Phys Rev B, 2008,77(19):195102(1-13).
[14] WANG F G, VALENTIN C D, PACCHIONI G. Doping of WO3 for photocatalytic water splitting: hints from density functional theory [J]. J Phys Chem C, 2012,116(16):8901-8909.
[15] COLE B, MARSEN B, MILLER E, et al. Evaluation of nitrogen doping of tungsten oxide for photoelectrochemical water splitting[J]. J Phys Chem C, 2008,112(13):5213-5220.
[16] MARSEN B, MILLER E, PALUSELLI D. Progress in sputtered tungsten trioxide for photoelectrode applications[J]. Int J Hydrogen Energy, 2007,32(15):3110-3115.
[17] SUN Y P, MURPHY C J, REYES-GIL K R, et al. Photoelectrochemical and structural characterization of carbon-doped WO3 films prepared via spray pyrolysis[J]. Int J Hydrogen Energy, 2009,34(20):8476-8484.
[18] KRESSE G, HAFNER J. Ab initio molecular dynamics for liquid metals[J]. Phys Rev B, 1993,47(1):558-561.
[19] KRESSE G, FURTHMLLER. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. J Phys Rev B, 1996,54(16):11169-11186.
[20] LOOPSTRA B, RIETVELD H. Structure of some alkaline-earth metal uranates[J]. Acta Crystallogr B, 1969,25(4):787.
[21] WIJS G A, BOER P K, GROOT R A, et al. Anomalous behavior of the semiconducting gap in WO3 from first-principles calculations[J]. Phys Rev B, 1999,59(4):2684-2693.
[22] VENKATESAN M, FITZGERALD C B, COEY J M D. Thin films: unexpected magnetism in a dielectric oxide [J]. Nature, 2004,430(7000):630-630.
[23] IVANOVSKII A L. Magnetic effects induced by sp impurities and defects in nonmagnetic sp materials [J]. Phys Usp, 2007,50(10):1031-1052.
[24] VOLNIANSKA O, BOGUSLAWSHI P. Magnetism of solids resulting from spin polarization of p orbitals[J]. J. Phys Condens Matter, 2010,22(7):073202.
[25] CUI X Y, MEDVEDEVA J E, DELLEY B, et al. Role of embedded clustering in dilute magnetic semiconductors: Cr Doped GaN [J]. Phys Rev Lett, 2005,95(4):256404.
[26] SAKONG S, KRATZER P. Density functional study of carbon doping in ZnO [J]. Semicond Sci Technol, 2011,26(1):014038.
[27] LARNBERT-MAURIAT C, OISON V, SAADI L, et al. Ab initio study of oxygen point defects on tungsten trioxide surface[J]. Surf Sci, 2012,606(1-2):40-45.
[28] YAN Z Z, LIU Q, JU L Q. Effects of lanthanide doping on electronic structures and optical properties of anatase TiO2 from density functional theory calculations [J]. J Phys D:Appl Phys, 2008,41(8):85417.
[29] TANG J, YE J. Photocatalytic and photophysical properties of visible-light-driven photocatalyst ZnBi12O20[J]. Chem Phys Lett, 2005,410(1-3):104-107.
(编辑 陈笑梅)
