肺腺癌吸烟相关甲基化模式识别分类模型及特征基因的识别研究
2016-02-16王世祥
王世祥 张 飞 王 玲 宋 凯,3*
1(天津大学化工学院,天津 300072)2(大连医科大学附属第一医院肿瘤科,辽宁 大连 116011)3(德克萨斯大学西南医学中心,美国 达拉斯 75235)
肺腺癌吸烟相关甲基化模式识别分类模型及特征基因的识别研究
王世祥1张 飞1王 玲2宋 凯1,3*
1(天津大学化工学院,天津 300072)2(大连医科大学附属第一医院肿瘤科,辽宁 大连 116011)3(德克萨斯大学西南医学中心,美国 达拉斯 75235)
吸烟是导致肺癌的一个重要诱导因素,从全基因组基因甲基化水平出发,利用生物信息学方法,通过建立对当前吸烟/不吸烟样本的模式识别分类模型,识别甲基化特征基因,为揭示不吸烟肺癌患者的患病机理奠定基础。为避免甲基化微阵列数据超高维小样本、高噪声、高相关性以及信息饱和现象淹没真正的特征基因,首次采用迭代多重筛选方法,分别从显著性差异、与基因表达水平的关系、生物功能、分类重要性等多个角度对全基因组甲基化数据进行多步筛选,从而识别吸烟相关特征基因。以TCGA数据库中127个肺腺癌样本为训练集,64个EDRN肺腺癌样本为独立测试集,最终确定了48个关键基因。相应模式识别模型对训练集精度达到87.5%(敏感性、特异性分别为87.2%和87.8%),独立测试集分类精度达到76.4%(敏感性、特异性分别为80.2%和73.6%)。交叉研究表明,其中17个基因对癌症发展的重要性已经在其他研究中有所证实,进一步的研究则证明其甲基化的重要性。同时,KEGG和IPA对特征基因在基因调控网络和代谢通路水平的分析表明,特征基因与癌症的发展以及生物功能、细胞发育等都有着密切的联系。
肺腺癌;甲基化数据;吸烟史;模式识别;分类
引言
吸烟是少数已知癌症的相关危险因素之一,与肺癌、膀胱癌、食管癌等癌症均有不同程度的联系[1],尤其是吸烟与肺癌的联系更是广为人知。然而,最近的统计研究表明,25%的肺癌患者并非吸烟所致。不吸烟患者肺癌的致死率高居第7位,甚至超过宫颈癌、胰腺癌和前列腺癌。
几乎所有肺癌的主要组织学子类均与吸烟有一定的联系,然而相比之下小细胞肺癌和肺鳞状细胞癌的联系则更为紧密[2]。与此不同,肺腺癌在非吸烟患者中的比例较大[3-4]。虽然肺癌的地域分布不同,但全球的统计数据表明,肺鳞状细胞癌的患者数量在逐年减少,而肺腺癌的患者数量却在激增[5]。其中,不吸烟肺腺癌患者的人数也在相应地逐年增长。为此,从多种角度研究吸烟史对癌症发生和发展的影响,找出不吸烟肺癌患者的发病机理,并开发相应的有针对性的治疗方案,则成为目前癌症研究领域的主要热点之一[6]。
微阵列技术等高通量基因测序技术及生物信息学的快速发展,为从基因组水平大规模地筛选吸烟相关致癌基因提供了必要的手段[7]。近年来,基于此种技术的研究结果表明,吸烟和不吸烟肺腺癌患者癌症的起源不同。在吸烟患者中,eGFR蛋白和细胞增殖标志物Ki-67的表达水平明显偏高;而在不吸烟患者中,AKT1和p27的表达水平则明显偏高。
随着高通量的DNA甲基化检测技术的出现,DNA甲基化的生物信息学研究得到了很大的发展。研究疾病特别是癌症中DNA甲基化的特点和规律,发现与特定癌症相关的甲基化生物标记,成为DNA甲基化研究中的热点问题。
近来的研究表明,DNA甲基化水平的改变在肿瘤发生和发展中起重要作用,可以通过重新激活沉默的致癌基因,引发相关癌症[8-9]。环境因素对DNA甲基化水平也有很大影响,尤其是长期吸烟或者是暴露在二手烟环境当中,都有可能在很大程度上改变DNA的甲基化水平[10]。例如,CDKN2A基因的甲基化在细胞周期调控中扮演重要角色,通常被认为是肺癌的潜在生物标志物,相比不吸烟样本,其异常高的甲基化水平在吸烟样本中更为常见[11]。Liu等发现,对于p16和MGMT两个基因,吸烟样本启动子区域甲基化的频率要显著高于不吸烟样本的情况[12]。Wu等的研究表明,hMLH1基因的甲基化在非小细胞肺癌的发展中起到重要作用,同时也是影响淋巴转移的重要因素[13]。
本研究以基因甲基化水平微阵列数据为主,利用先进的模式识别技术,对吸烟与不吸烟的肺腺癌样本进行分类;通过对模型参数的优化,识别具有最优分类能力的最小基因集(即甲基化特征基因),揭示肺腺癌吸烟与不吸烟患者在基因甲基化水平上的异同,为进一步解释不吸烟肺癌患者的患病机理提供理论依据。
为克服全基因组数据超高维高噪声小样本特性对机器学习算法性能的影响,防止信息饱和(即少数重要基因信息淹没于数万基因所含的噪声中)现象,笔者创新性地组合应用3种特征基因筛选方法,并通过迭代降维技术递归筛选真正的特征基因。
在研究中,以癌症基因组图谱(the cancer genome atlas, TCGA)肺腺癌甲基化水平数据为训练样本,以EDRN(the early detection research network)肺腺癌甲基化水平数据为独立测试样本。从2万多个基因相应的48万多个探针数据中,最终筛选出48个基因。仅利用这48个基因的甲基化水平数据,即可将病人样本分为“吸烟/不吸烟”两类。其中,TCGA训练样本的分类精度高达87.5%,EDRN独立测试集的分类精度也高达76.4%。相应的代谢和基因调控网络分析充分证明了48个特征基因在肺癌代谢通路水平上的强相关性。同时,EDRN数据与TCGA数据检测平台的不同也充分说明了特征基因在不同平台数据应用中的鲁棒性。
1 数据和方法
1.1 数据样本
1.1.1 TCGA数据
甲基化数据为癌症基因组图谱(the cancer genome atlas, TCGA, https://tcga-data.nci.nih.gov/ tcga/tcgaHome2.jsp)数据库中的第三水平(level 3)数据,即经过TCGA初步预处理的数据。数据检测平台为Illumina Infinium Human Methylation 450。
删除所有吸烟史信息缺失的样本后共获得127个甲基化数据样本,其中73个当前吸烟样本(当前吸烟或者戒烟少于12个月)和54个不吸烟样本(终身吸烟数量不超过100根),相应的临床信息见表1。

表1 相应数据临床信息汇总Tab.1 Clinical information of the corresponding data
1.1.2 EDRN数据
采用GEO公共数据平台(http://www.ncbi.nlm.nih.gov/geo/)下载的EDRN甲基化数据作为独立测试集[14],该数据检测平台为Illumina Infinium Human Methylation 27。
同样,删除吸烟史缺失的样本,最后保留64个甲基化样本(其中34个当前吸烟样本和30个不吸烟样本),相应的临床信息见表1。
1.2 数据预处理
近来的研究表明,影响基因表达水平的甲基化主要集中在相应的启动子区域[15-16]。为此,首先从48万(TCGA数据)和2.7万(EDRN)个甲基化水平检验探针数据中筛选位于基因启动子区域的探针数据,以其均值作为相应基因的甲基化水平。之后删除所有在X、Y染色体上的基因数据以及所有样本中全为空值的基因数据,最后求取两组数据平台的基因交集,最终剩余13 564个基因。
利用TCGA数据作为训练集,EDRN数据作为测试集。为了克服数据之间的不平衡性,针对TCGA数据,在吸烟样本和不吸烟样本中,各随机抽取相同数目样本(50个)组成训练集;而对于EDRN数据,同样随机抽取相同数目样本(30个)组成测试集,利用5重交叉验证的方法对分类模型进行优化。
1.3 特征基因识别
如前诉述,本研究中共有13 564个备选基因,特征基因通常仅有几十个。为防止信息饱和现象淹没真正的特征基因,同时克服微阵列数据的超高维小样本特性以及基因变量间的多重共线性对模式识别分类模型精度的负面影响,笔者首次采用迭代多重筛选方法,对全基因组数据进行多步筛选。
筛选方法1:SAM筛选。基因表达差异显著性分析(SAM)是一种常用的微阵列数据预处理方法,常用于高维基因数据的初步筛选。SAM通过多重基因特异性检验,识别具有显著差异的基因;通过错误发现率(FDR)算法,控制多重检验的错误率[17-19]。本研究选用SAM算法筛选(初步筛选3000个基因),作为候选基因的第一部分。
筛选方法2:相关性筛选。大量研究表明,DNA甲基化能引起染色质结构、DNA构象、DNA稳定性及DNA与蛋白质相互作用方式等的改变,从而影响基因表达[20]。相比之下,能够影响基因表达水平的甲基化具有更加显著的生物学意义,对癌症的发展能起到更加关键的作用。因此,筛选甲基化水平与基因表达水平具有明显相关性(相关系数的绝对值大于0.5)的基因作为候选基因集的第二部分。
筛选方法3:PLS综合筛选。为突出24个已知重要基因(附表S1已列出24个基因的相关信息)的作用,将其与前两步筛选的候选基因集合并后采用PLS算法重新筛选。合并后基因交集共包含3 427个基因,相比13 564个基因,候选基因数量大大减少,有效克服了信息饱和现象对已知重要基因的削弱。另外,SAM虽然能够克服常规显著性检验的局限性,并在一定程度上限制了错误发现率,但毕竟属于单变量分析方法,无法克服噪声及变量相关性的影响,因此只适合于基因变量的初步筛选。PLS(部分最小二乘)算法通过提取与原始变量线性相关的互相正交的潜变量[21],将原始高维样本压缩至低维空间进行模式识别和回归分析,因此能够有效地克服数据中噪声和多重相关性等问题,在生物信息学领域得到越来越广泛的应用。为此,本研究采用PLS筛选最终的特征基因。通过对模式识别模型分类精度的递归迭代优化,具有最优分类精度的最小基因集即为最终的特征基因集。
具体流程如图1所示。SAM、PLS均在R语言环境下运行,相应细节可参见实验室网站http://www.csssk.net提供的附件支持材料。
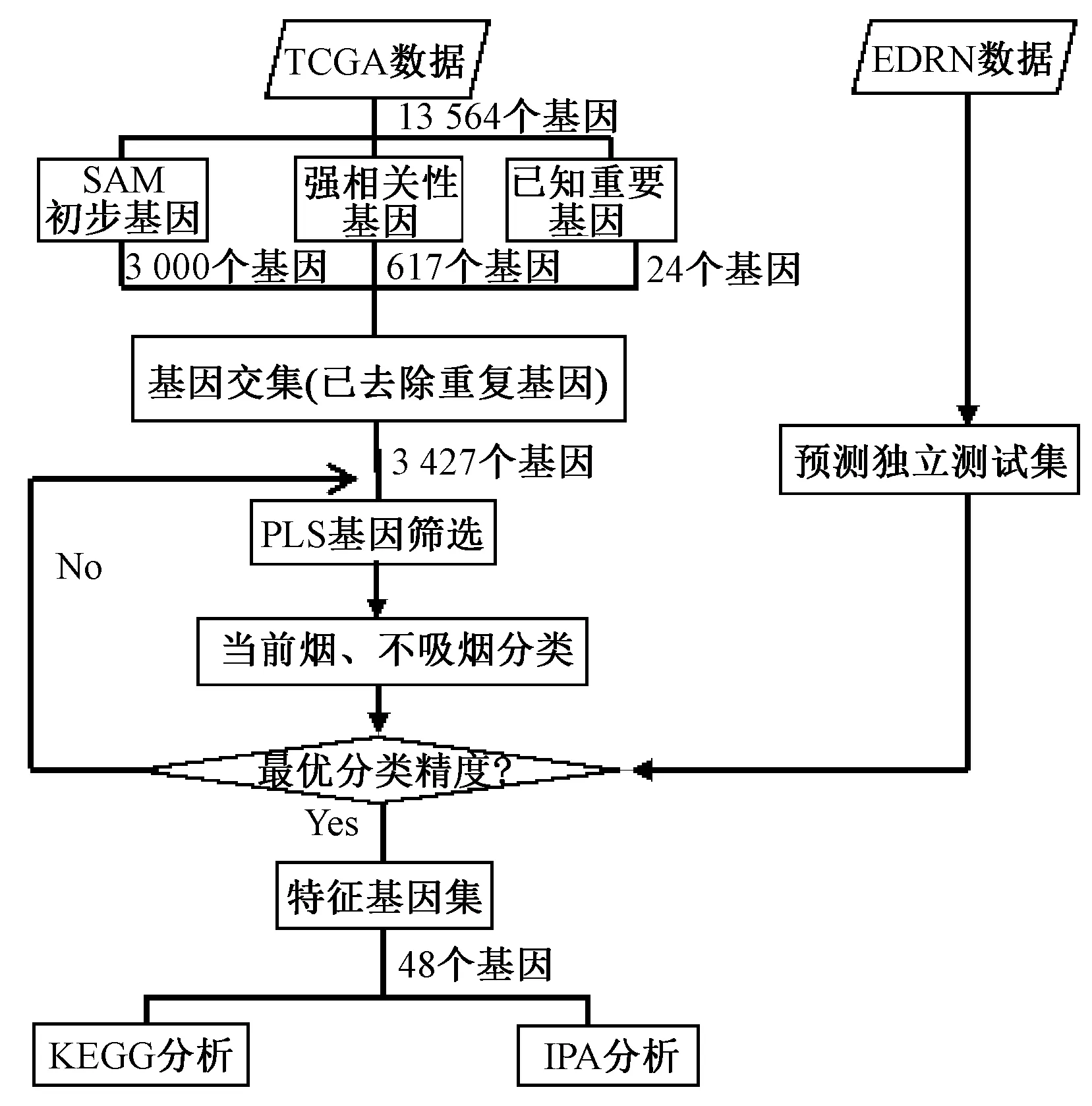
图1 吸烟相关甲基化模式识别分类模型研究及特征基因识别流程Fig.1 The flow chart of genome-wide smoke related methylation signature genes identification
1.4 分类模型评价
为了评价分类模型的性能,采用了准确率(accuracy)、特异性(specificity)和敏感性(sensitivity)3个指标。
(1)
(2)
(3)
在研究中,正样本为不吸烟样本,负样本为吸烟样本。true positive(TP)表示不吸烟样本正确分类的个数,false positive(FP)表示不吸烟样本被误判为吸烟样本的个数;true negative(TN)表示吸烟样本正确分类的个数,false negative(FN)表示吸烟样本被误判为不吸烟样本的个数。
sensitivity(SN)表示不吸烟样本正确分类的比率,specificity(SP)表示吸烟样本正确分类的比率,accuracy(ACC)表示所有样本正确分类的比率。
所有算法均在R环境下运行和优化。相应详细介绍和程序可参见附件或访问实验室网站http://www.csssk.net。
2 结果
在本研究中,共有13 564个备选基因,采用迭代多重筛选方法,对全基因组数据进行多步筛选,最终剩余48个特征基因。作为候选基因集的初步筛选,笔者创新性地结合多种方法SAM筛选,通过错误发现率(FDR)算法控制多重检验的错误率,筛选出3 000个基因;基于t-test的相关性筛选,选出617个强相关性基因;加之其他文献中提及的已经被实验所验证的24个已知重要基因,共剩余3 427个基因(已去除重复基因)组成候选基因集。最后利用PLS算法,通过对模式识别模型分类精度的递归迭代优化的方法,逐个删减基因,并确保分类精度达到最优,最终剩余48个基因。
2.1 模式识别模型结果
本研究的目标是识别能够区别吸烟/不吸烟肺腺癌患者的甲基化特征基因,揭示不吸烟患者的患病机理,为开发更有针对性的治疗方法奠定基础。为此,所识别的相关甲基化特征基因集必须具有足够高的分类能力,才能有效证明其代表性:仅采用特征基因的甲基化水平,即可对病人样本按照吸烟史进行分类(当前吸烟和不吸烟两类)。本研究所识别的48个甲基化特征基因,相应模式识别分类模型的分类结果如表2所示。由结果可知,仅采用48个特征基因的甲基化水平,TCGA训练集的精度即可高达87.5%,EDRN独立测试集的精度为76.4%。
表2 甲基化特征基因模式识别分类结果(当前吸烟/不吸烟)Tab.2 The results of methylation signature genesidentification
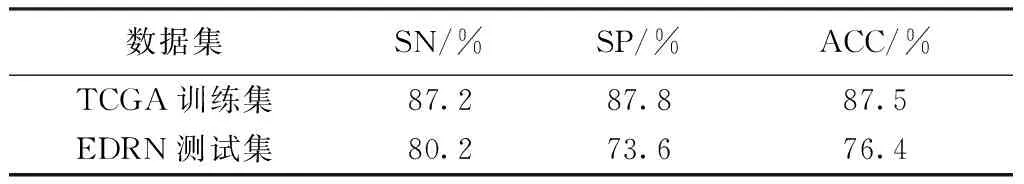
数据集SN/%SP/%ACC/%TCGA训练集87.287.887.5EDRN测试集80.273.676.4
此外,无论是对训练样本还是对独立测试集样本,模式识别的特异性和敏感性均非常接近。对EDRN而言,其特异性为73.6%,敏感性为80.2%,相差仅为6.6%,虽然大于TCGA训练集的相应偏差,但足以说明模式识别模型具有良好的平衡性和可靠性。
2.2 甲基化特征基因
所识别的48个特征基因,按其在不吸烟样本中甲基化数据的中位数进行排序,得到如图2所示的箱线图。

图2 特征基因箱线图(特征基因按其在不吸烟样本中甲基化数据的中位数进行排序,颜色随甲基化水平递增而逐渐加深)。(a)不吸烟样本(n=54);(b)吸烟样本(n=73)Fig.2 Boxplot of signature genes(Signature genes was sorted by median of methylation levels in the never smokers. Color deepens with increasing methylation level)(a)Never-smokers(n=54);(b)Current-smokers(n=73)

KEGGID通路名称P值通路包含的基因hsa05218Melanoma2.529×10-4EGFR,KRAS,MET,FGF11,MDM2hsa05200Pathwaysincancer4.339×10-4EGFR,RET,KRAS,MET,PAX8,FGF11,MDM2,STAT1hsa05214Glioma0.0027EGFR,KRAS,CALML3,MDM2hsa05216Thyroidcancer0.0073RET,KRAS,PAX8hsa04144Endocytosis0.0086EGFR,RET,ERBB3,MET,MDM2hsa05219Bladdercancer0.0151EGFR,KRAS,MDM2hsa05223Non-smallcelllungcanc-er0.0243EGFR,FHIT,KRAShsa05212Pancreaticcancer0.0413EGFR,KRAS,STAT1hsa04020Calciumsignalingpath-way0.0434EGFR,CALML3,ERBB3,HTR2Bhsa05210Colorectalcancer0.0545EGFR,KRAS,METhsa04012ErbBsignalingpathway0.0580EGFR,KRAS,ERBB3hsa04540Gapjunction0.0604EGFR,KRAS,HTR2Bhsa05215Prostatecancer0.0604EGFR,KRAS,MDM2hsa04912GnRHsignalingpathway0.0716EGFR,KRAS,CALML3
可以看出,在吸烟与不吸烟样本中,特征基因的甲基化水平有明显差异,吸烟样本中甲基化水平更加分散,而在不吸烟样本中甲基化水平更紧密。例如,GLDC、CYBA、CD40、WBSCR17、PXMP4等基因,说明吸烟作为一个危险因素会对甲基化水平带来一定影响。同时,对于SRGN、GTSF1、WBSCR17、OLFM4、CD40、GPR152、RNASE6、MMP25、CA6、PXMP4等基因,其吸烟与不吸烟样本甲基化水平具有显著差异(P<0.001),而对于基因DUSP6、SULT4A1、EGFR、B4GALNT4、MET、RET、MMD、CHRNA5,其P>0.5,详细可见附件(下载网址为http://www.csssk.net)中的附表S2。但根据其对模式识别模型的贡献率可知,如基因B4GALNT4、DUSP6的贡献率均排在前7位,并且从功能分析可知,如EGFR、MET、RET等基因,其甲基化水平对吸烟导致肺癌发生和发展的机理至关重要。
2.3 特征基因的基因通路
2.3.1 KEGG分析
KEGG(Kyoto encyclopedia of genes and genomes)是系统分析基因功能、基因组信息的数据库,它有助于研究者在代谢水平上对特征基因作为一个整体网络进行研究,是进行生物体内代谢分析、代谢网络研究的强有力工具。David分析平台的KEGG分析软件的链接为http://david.abcc.ncifcrf.gov/。
对于48个ME特征基因,共有14条重要的KEGG代谢通路,具体结果如表3所示。
可以看出,它们中的大多数都与癌症有关。其中,非小细胞肺癌代谢通路(hsa05223)也包含在内,P<0.05。除此之外,参与这些代谢通路的基因,如EGFR、 KRAS、MDM2、FHIT、RET等,都已被证实在肺腺癌的发生发展中起到十分重要的作用,证明了本研究所确定出的特征基因的重要性。
2.3.2 IPA分析
IPA(ingenuity pathway anaylsis)分析用来揭示已确定的甲基化特征基因的基本网络功能关系,基因集共包含48个基因,共11个基因调控网络与这些基因具有直接关系。其中,两个主要调控网络如图3所示,其他信息汇总于表4。这两个调控网络中所包含的大部分基因都与癌症密切相关,在癌症的发展过程中扮演着重要的角色。另外,相应调控网络的其他生物功能还包括细胞生长及增殖(cellular growth and proliferation)、细胞发育(cellular development)、细胞间信号传导和相互作用(cell-to-cell signaling and interaction)、细胞功能和维护(cellular function and maintenance)等。

图3 ME特征基因IPA主要调控网络。(a)调控网络1;(b)调控网络2Fig.3 IPA network of signature genes.(a)IPA network 1;(b)IPA network 2

编号基因位置编号基因位置编号基因位置1GSTM11p13.317WBSCR177q11.2333ERBB312q132LCK1p34.318MET7q3134GTSF112q13.23C1orf641p36.1319C7orf457q32.235MDM212q14.3-q154CA61p36.220AKR1B107q3336DUSP612q22-q235ANKRD451q25.121NAT28p2237OLFM413q21.16GNLY2p11.222ZNF5728q24.1338RNASE614q11.27ALK2p2323GLDC9p2239CHRNA515q248PAX82q12-q1424SPAG610p12.240MMP2516p13.39STAT12q32.225CALML310p15.141CYBA16q2410HTR2B2q36.3-q37.126PPYR110q11.242FGF1117p13.111FHIT3p14.227RET10q11.243MMD17q12UCHL14p1428SRGN10q22.144ZFP2819q13.4313PCDHB115q3129MGMT10q2645CPXM120p13-p12.314LEAP25q31.130B4GALNT411p15.546PXMP420q11.2215TCP116p21.3-p21.231GPR15211q13.147CD4020q12-q13.216EGFR7p1232KRAS12p12.148SULT4A122q13.2-q13.31
在研究中,通常认为一个基因有5或5条及以上直接连接的为Hub基因。本研究中的48个基因共有6个Hub基因:EGFR、ERBB3、STAT1、MET、MDM2和CD40。
1)EGFR (epidermal growth factor receptor) 有13条连接。EGFR基因是上皮生长因子细胞增殖和信号传导的受体,其突变或过表达一般会引发肿瘤。研究表明,在许多实体肿瘤中存在EGFR的高表达或异常表达[22]。
2)ERBB3 (Erb-B2 Receptor Tyrosine Kinase 3) 有8条连接。它编码表皮生长因子受体(EGFR)家族受体酪氨酸激酶,此基因的过表达已经出现在许多癌症中[23]。
3)STAT1 (signal transducer and activator of transcription 1) 有6条连接。STAT1基因可以通过控制免疫系统,促进肿瘤免疫监视能力,提高免疫系统识别、杀伤,并及时清除体内突变细胞,防止肿瘤发生,它在肿瘤起始阶段发挥重要作用[24]。
4)MET (METProto-Oncogene, Receptor Tyrosine Kinase)有6条连接。它在非小细胞肺癌的细胞株和肿瘤组织中均有过表达现象,并且c-Met在非小细胞肺癌中有重要的生物学意义[25]。
5)MDM2 (murine double minute 2) 有6条连接。MDM2基因参与调节细胞增殖和凋亡相关的信号通路,在多种恶性肿瘤中都发生异常改变,其过表达促进了肿瘤的发生和发展,与恶性肿瘤的发展密切相关[26]。
6)CD40 (CD40 Molecule, TNF Receptor Superfamily Member 5) 有5条连接。CD40在肿瘤细胞表面表达和肿瘤的转移扩散之间存在显著的统计学相关性,而肺癌中CD40的表达可能在转移扩散中起到关键作用,并且可作为预后标志和晚期疾病的指标[27]。
3 讨论
通常意义下,“不吸烟”指患者个人终生接触的香烟数量少于100根,“曾经吸烟”者指患者个人戒烟超过至少12个月,“当前吸烟者”指当前正在吸烟或者戒烟不超过12个月的患者。除此以外,日本等少数国家对吸烟史的定义则不尽相同。鉴于对吸烟史的不同定义及患者对此的不同理解,“不吸烟”患者样本中难免会有部分“曾经吸烟”患者样本。另外,患者戒烟的时间越长,包括基因组突变特性的各项基因组特征越来越接近于“不吸烟”患者。相反,长期暴露在二手烟或者相应环境污染下的“不吸烟”患者,其各项基因组特征则更接近于“当前或者曾经”吸烟者。由此带来的训练样本类别误差是不能避免的,基于此的模式识别分类精度则不可能高达100%。但是,由表2可以看出,本研究的模式识别模型仍能以87.5%的精度识别训练样本,且更以76.4%的精度识别EDRN独立测试集,充分说明了模式识别模型和特征基因集的准确性及代表性。
更为重要的是,如前所述,TCGA训练集数据的测试平台为Illumina Infinium Human Methylation 450,而EDRN的测试平台为Illumina Infinium Human Methylation 27。表2所示的结果则进一步证明了本研究的模式识别分类模型和特征基因集对不同平台数据的鲁棒性。
如前所述,图2中的基因DUSP6、SULT4A1、EGFR、B4GALNT4、MET、RET、MMD、CHRNA5,其P>0.5,在两类样本中差异并不显著,但根据其对模式识别模型的贡献率可知,基因B4GALNT4、DUSP6的贡献率均排在前7位,并且从功能分析可知,如EGFR、MET、RET等基因,其甲基化水平对吸烟导致肺癌发生和发展的机理至关重要。为了进一步验证这些基因对模式识别模型的重要性,从48个特征基因中去除这些基因,仅利用显著差异基因对当前吸烟/不吸烟样本进行模式识别分类,结果表明,对于TCGA训练集,模式识别精度为82.3%,而EDRN独立测试集的精度为70.5%,与训练集87.5%、独立测试集76.4%的最高精度相比有所下降,从而更加证明了这些基因对模式识别模型的显著作用。
此外,48个特征基因中某些基因的重要性已经被其他研究所证实。
1)Nakajima等已经证实,在吸烟样本中GSTM1基因表达缺失,会导致GSTM1-1酶和GSTM3-3酶的欠表达,使得毒性的降解不能正常进行,相应地使活跃在烟草烟雾中的致癌物质增加,这种致癌物质代谢的不平衡造成了肺癌的发生[28]。在本研究中,吸烟样本GSTM1甲基化与表达水平的相关系数是-0.877,呈现明显的负相关,高甲基化导致了基因表达的缺失。
2)Liu等发现,与从不吸烟样本相比,在吸烟样本中p16和MGMT两个基因启动子区域甲基化的频率显著偏高[12],说明非小细胞肺癌的发生与吸烟之间有很强的相关性。在本研究中,MGMT基因启动子区域甲基化的频率也显著偏高。
3)KRAS基因的突变也非常常见,存在于约30%的肺腺癌和约5%的鳞状细胞肺癌中[29],在肺癌进展中起了重要的作用。EGFR突变激活的PI3K-AKT 和 RAS-MEK-ERK信号对癌细胞的生长、生存和迁移起至关重要的作用[30-31]。这两个基因都是肺癌的重要生物标记。图4显示了KRAS与EGFR基因在突变样本与未突变样本中的甲基化水平,两基因在突变样本与未突变样本中都有显著差异性,P值分别为4.44×10-16和4.43×10-16。由此可见,其甲基化水平与突变之间具有很强的相关性。

图4 KRAS与EGFR基因蜂群图。(a)KRAS;(b) EGFRFig.4 Beeswarm plot of KRAS and EGFR genes.(a)KRAS;(b) EGFR
4)高表达的基因(如MDM2)可在肿瘤的发生、发展过程中起到关键作用,这是因为它利用双分染色体的能力,迅速繁殖并显著增加这些细胞的致癌能力[32]。MDM2基因表达水平的不同可能与其甲基化水平的不同有关,本研究中MDM2在吸烟样本中的甲基化水平更低,这可能造成了MDM2在吸烟样本中的异常表达,导致癌症的发生。
5)脆性组氨酸三联(FHIT)基因的功能缺失,已被证明是已知识别肺癌的关键生物标记[33-34]。
除此之外,在48个甲基化特征基因中,已知的与非小细胞肺癌密切相关的关键基因还包括LCK、NAT2、CHRNA5、AKR1B10、DUSP6、MMD、STAT1、RET、ERBB3、ALK、MET,占所识别全部48个特征基因的35.4%,都已被相关的实验所验证。由此可以看出,本研究所提出的识别肺腺癌吸烟相关特征基因的方法具有可行性,同时也证明了所确定基因集的可信度,为指导临床个性化治疗提供了依据。
4 结论
本研究创新性地采用多重迭代筛选方法,分别从显著性差异、与基因表达水平的关系、生物功能、分类重要性等多个角度,对全基因组甲基化数据进行多步筛选,使结果更加全面可信。识别出的48个吸烟相关特征基因,存在着显著的生物学意义,为揭示吸烟与肺腺癌的关系以及不吸烟患者的患病机理提供了依据,同时为开发更有针对性的治疗方法奠定了基础。
[1] Figueroa JD, Han SS, Garcia-Closas M, et al. Genome-wide interaction study of smoking and bladder cancer risk [J]. Carcinogenesis, 2014, 35(8): 1737-1744.
[2] Figueroa JD, Han SS, Garcia-Closas M, et al. Genome-wide interaction study of smoking and bladder cancer risk [J]. Carcinogenesis, 2014, 35(8): 1737-1744.
[3] Toh CK, Gao F, Lim WT, et al. Never-smokers with lung cancer: epidemiologic evidence of a distinct disease entity [J]. Journal of Clinical Oncology, 2006, 24(15): 2245-2251.
[4] Kiyohara C, Wakai K, Mikami H, et al. Risk modification by CYP1A1 and GSTM1 polymorphisms in the association of environmental tobacco smoke and lung cancer: a case-control study in Japanese nonsmoking women [J]. International Journal of Cancer, 2003, 107(1): 139-144.
[5] Gabrielson E. Worldwide trends in lung cancer pathology [J]. Respirology, 2006, 11(5): 533-538.
[6] Radzikowska E, Glaz P, Roszkowski K. Lung cancer in women: age, smoking, histology, performance status, stage, initial treatment and survival [J]. Annals of Oncology, 2002, 13(7): 1087-1093.
[7] Allison DB, Cui X, Page GP, et al. Microarray data analysis: from disarray to consolidation and consensus [J]. Nature Reviews Genetics, 2006, 7(1): 55-65.
[8] Kim SC, Jung Y, Park J, et al. A high-dimensional, deep-sequencing study of lung adenocarcinoma in female never-smokers [J]. PLoS ONE, 2013, 8(2): e55596.
[9] Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data [J]. Bioinformatics, 2014, 30(15): 2114-2120.
[10] Lee KW, Pausova Z. Cigarette smoking and DNA methylation [J]. Frontiers in Genetics, 2013, 4(1): 132-142.
[11] Selamat SA, Galler JS, Joshi AD, et al. DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma [J]. PLoS ONE, 2011, 6(6): e21443.
[12] Liu Yang, Lan Qing, Siegfried JM, et al. Aberrant promoter methylation of p16 and MGMT genes in lung tumors from smoking and never-smoking lung cancer patients [J]. Neoplasia, 2006, 8(1): 46-51.
[13] Wu Fang, Lu Min, Qu Lu, et al. DNA methylation of hMLH1 correlates with the clinical response to cisplatin after a surgical resection in non-small cell lung cancer [J]. International Journal of Clinical and Experimental Pathology, 2015, 8(5): 5457-5463.
[14] Selamat SA, Chung BS, Girard L, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression [J]. Genome Research, 2012, 22(7): 1197-1211.
[15] Jones PA, Laird PW. Cancer-epigenetics comes of age [J]. Nature Genetics, 1999, 21(2): 163-167.
[16] Jones PA. The DNA methylation paradox[J]. Trends in Genetics, 1999, 15(1):34-37.
[17] George G, Raj VC. Review on feature selection techniques and the impact of SVM for cancer classification using gene expression profile [J]. International Journal of Computer Science & Engineering Survey, 2011, 2(3): 42-55.
[18] Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response [J]. Proceedings of the National Academy of Sciences, 2001, 98(9): 5116-5121.
[19] Zhang Chunying, Girard L, Das A, et al. Nonlinear quantitative radiation sensitivity prediction model based on NCI-60 cancer cell lines [J]. The Scientific World Journal, 2014, 2014(5): 602-612.
[20] Phillips T. The role of methylation in gene expression [J]. Nature Education, 2008, 1(1): 116-121.
[21] Abdi H. Partial least square regression (PLS regression) [J]. Encyclopedia for Research Methods for the Social Sciences, 2003, 6(4): 792-795.
[22] Sun Guangyuan, Liu Bing, He Jin, et al. Expression of EGFR is closely related to reduced 3-year survival rate in Chinese female NSCLC [J]. Medical Science Monitor, 2015, 21(1): 2225-2231.
[23] Bublil EM, Yarden Y. The EGF receptor family: spearheading a merger of signaling and therapeutics [J]. Current Opinion in Cell Biology, 2007, 19(2): 124-134.
[24] Koromilas AE, Sexl V. The tumor suppressor function of STAT1 in breast cancer [J]. Jak-Stat, 2013, 2(2): 1-5.
[25] Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer [J]. Cancer Research, 2005, 65(4): 1479-1488.
[26] Liu G, Wheatley-Price P, Zhou Wei, et al. Genetic polymorphisms of MDM2, cumulative cigarette smoking and nonsmall cell lung cancer risk [J]. International Journal of Cancer, 2008, 122(4): 915-918.
[27] Sabel MS, Yamada M, Kawaguchi Y, et al. CD40 expression on human lung cancer correlates with metastatic spread [J]. Cancer Immunology Immunotherapy, 2000, 49(2): 101-108.
[28] Nakajima T, Elovaara E, Anttila S, et al. Expression and polymorphism of glutathione S-transferase in human lungs: risk factors in smoking-related lung cancer [J]. Carcinogenesis, 1995, 16(4): 707-711.
[29] Korpanty GJ, Graham DM, Vincent MD, et al. Biomarkers that currently affect clinical practice in lung cancer: EGFR, ALK, MET, ROS-1, and KRAS [J]. Frontiers in Oncology, 2014, 4(1): 204-211.
[30] Heist RS, Engelman JA. SnapShot: non-small cell lung cancer [J]. Cancer Cell, 2012, 21(3): 448-448.e2.
[31] Wistuba II, Gazdar AF. Lung cancer preneoplasia [J]. Annu Rev Pathol Mech Dis, 2006, 1(1): 331-348.
[32] Sanborn JZ, Salama SR, Grifford M, et al. Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons [J]. Cancer Research, 2013, 73(19): 6036-6045.
[33] Westra WH, Baas IO, Hruban RH, et al. K-ras oncogene activation in atypical alveolar hyperplasias of the human lung [J]. Cancer Research, 1996, 56(9): 2224-2228.
[34] Sozzi G, Pastorino U, Moiraghi L, et al. Loss of FHIT function in lung cancer and preinvasive bronchial lesions[J]. Cancer Research, 1998, 58(22):5032-5037.
Genome-Wide Smoke Related Methylation Signature Genes Identification for Lung Adenocarcinomas
Wang Shixiang1Zhang Fei1Wang Ling2Song Kai1,3*
1(SchoolofChemicalEngineeringandTechnology,TianjinUniversity,Tianjin300072,China)2(FirstAffiliatedHospitalofDalianMedicalUniversity,Dalian116011,Liaoning,China)3(UniversityofTexasSouthwesternMedicalCenter,Dallas75235,USA)
To understand the biological mechanism of never smoker lung adenocarcinomas, we focused on the genome-wide methylation values (ME) to discover signature genes for the distinguishing of current/never smokers. In order to overcome the disadvantages of small-size-high-dimension, high noise and to overcome the predominate influence of the whole genome to the dozens of signature genes, a new integrative selection method was used iteratively to uncover the real signature genes. To do this, instead of using only one criteria for gene selection, we identified genes according to their significance test performance, the relationship between their methylation levels and expression levels, the biological function and the contribution to the current/never smoker classification. As a result, 48 genes were identified as ME smoke related signature genes based on the 127 lung adenocarcinoma samples downloaded from TCGA database. Then we used 64 EDRN lung adenocarcinoma samples as an independent validation set. Only using the methylation values of these 48 signature genes, the current/never smoker classification accuracy of TCGA training set is 87.5% (SN=87.2%, SP=87.8%) and for EDRN validation set is 76.4% (SN=80.2%, SP=73.6%), respectively. Cross-study proved the highly cancer related of 17 important genes in our 48 signature genes. Addition to these results, we proved the importance of their corresponding methylation values. The ingenuity pathway (IPA) and Kyoto encyclopedia of genes and genomes (KEGG) pathways analysis indicated the relationships among these genes on the genetic network level and pathway levels. They also indicated they are involved in the highly cancer-related pathways.
lung adenocarcinoma;methylation values;smoke exposure;pattern recognition;classification
10.3969/j.issn.0258-8021. 2016. 03.007
2015-10-12, 录用日期:2016-03-08
国家自然科学基金(31271351)
R318
A
0258-8021(2016) 03-0301-09
*通信作者(Corresponding author), E-mail:ksong@tju.edu.cn
