关于弯曲界面气液两相相平衡化学势相等热力学判据的讨论
2016-02-13肖赛君尹振兴
肖赛君 刘 健 尹振兴 章 俊
(安徽工业大学冶金工程学院,安徽马鞍山243002)
关于弯曲界面气液两相相平衡化学势相等热力学判据的讨论
肖赛君*刘 健 尹振兴 章 俊
(安徽工业大学冶金工程学院,安徽马鞍山243002)
弯曲界面气液两相相平衡时化学势相等的热力学判据见诸于大量教材和论文。本文对现有教材中关于该判据的推导过程进行了分析,认为该判据的证明过程存在问题。为解决该问题,本文从化学势的定义出发提供了一种化学势相等热力学判据的新证明过程。同时,本文还依据吉布斯界面热力学理论建立了另一种弯曲界面气液两相相平衡的热力学新判据,并以此为基础进行开尔文公式的推导。新判据直接从热力学第二定律出发,热力学意义明确,物理模型清晰,整个推导过程简洁明了。
弯曲界面气液两相平衡;化学势;开尔文方程
目前,国内外物理化学[1—8]与表面热力学教材[9—11]以及相关论文[12—14]均将化学势相等作为弯曲界面气液两相达到相平衡的热力学判据,并以该判据为基础,将液滴化学势转化为存在附加压力时的化学势进行开尔文方程的推导。该热力学判据最早由吉布斯提出[15],吉布斯建立该判据的假设前提是“与分界表面有关的那部分的能量和熵的改变,与均相部分的能量和熵的变化相比较,可以忽略不计”[15],即吉布斯忽略了表面相的能量,以不含表面相的体相的化学势相等作为相平衡的近似判据。国内教材中,弯曲界面气液两相相平衡化学势相等的热力学判据有严格推导,推导最早见于王竹溪先生所著《热力学》[16],在其随后的系列热力学专著中也均有介绍[17—20]。该推导方法已为大多数教材和论文所引用。
本文系统梳理了王竹溪先生关于弯曲界面气液两相化学势相等热力学判据的推导过程,分析表明,该判据的证明过程存在若干问题。为解决该问题,本文从化学势的定义出发提供了一种化学势相等热力学判据的新证明过程。同时,本文还依据吉布斯界面热力学理论建立了另一种弯曲界面气液两相相平衡的热力学新判据,并以此为基础进行了开尔文方程的推导。新判据直接从热力学第二定律出发,热力学意义明确,物理模型清晰,整个推导过程简洁明了,值得推广。
1 弯曲界面气液两相相平衡化学势相等热力学判据的推导
王竹溪先生所著系列热力学教材均对有弯曲界面时气液两相相平衡的热力学判据进行了推导,所得结论为:对于弯曲界面气液两相相平衡的条件是气体与液体的化学势相等。此时液体的化学势为附加压力下的化学势,附加压力的大小由拉普拉斯公式决定。以球状液滴为例,气液两相相平衡的热力学判据如式(1)所示。

关于式(1)的推导过程可参见王竹溪先生系列教材的“30.有曲面分界的平衡条件”一节。由于教材采用旧的热力学符号体系,本文将其“翻译”为新符号体系予以介绍。同时,为了使推导过程的物理模型清晰,让推导过程成为一个有机整体,本文在不改变原文意思的前提下做了适当调整。
首先建立如图1所示的物理模型。恒温恒容条件下,小液滴及其平衡气相由α相、β相和弯曲的界面相σ组成。在吉布斯界面模型中,界面相σ被认为是没有体积只有面积的几何相。
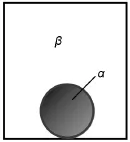
图1 球状液滴与气相的平衡示意图
为推导弯曲界面两相相平衡的热力学判据,针对图1所示气液两相平衡状态,教材[16—20]先后建立了两个热力学过程。
第一个热力学过程为假定气液两相之间没有物质交换,α相与β相的体积发生无限小变化dVα和dVβ,σ相的面积相应发生变化为dAS。对整个系统,依据吉布斯界面热力学基本方程,有式(2)。

已知恒温恒容且无非体积功时的热力学平衡判据为dA=0,即令式(2)等于零,有式(3)。

由于王竹溪先生所著教材对式(3)的推导没有说明其对应的热力学过程,关于上述第一个热力学过程的推导,本文主要采用内容与王竹溪先生教材相同的胡英先生《物理化学》教材[3]中的推导过程。
仍以图1所示恒温恒容条件下的气液两相平衡状态为对象,建立第二个热力学过程。假设液相α相的物质的量改变为dnαmol。dnαmol的物质全部进入β相,β相物质的量改变为dnβmol,则dnα+dnβ=0。依据吉布斯界面热力学基本方程,在此转移过程中,整个体系dA=0。
对于液相α,dAα=—pαdVα+μαdnα+σdAS;对于气相β,dAβ=—pβdVβ+μβdnβ。对于整个体系,dA=dAα+dAβ=0,将dAα与dAβ的具体表达式代入得式(4)。

将第一个热力学过程所得的式(3)代入式(4),则得式(5)。

将dnα+dnβ=0代入式(5),则得式(6)。

式(6)即图1所示球状液滴与气相平衡时,气体与液体化学势相等的热力学判据。第二个热力学过程的推导全部来自于王竹溪先生教材“30.有曲面分界的平衡条件”一节中,本文仅将旧的热力学符号体系进行了转换。
2 关于弯曲界面气液两相相平衡化学势相等热力学判据的讨论
2.1 关于该判据推导过程的讨论
(1)关于第一个热力学过程的分析讨论。
热力学过程的定量推导都应该建立在清晰合理的物理图像上,不清晰的物理图像往往导致过度复杂,甚至不知所云的数学推导。上述第一个热力学过程的假设条件是恒温恒容液滴体积增大dV过程中不存在外力做非体积功,即在没有外力做功条件下,球状液滴体积增大dVα。从热力学第一定律来考查该过程球状液滴的能量变化。在温度不变、没有外力做功的情况下,dAα=—pαdVα+σdAS,该式表明,液滴α相在没有外力做功的条件下,既对气体β相做体积功,同时自身的表面能还增加,这明显不符合能量守恒定律。所以,第一个热力学过程所得的结论是有问题的。
(2)关于第二个热力学过程的分析讨论。
按照第一个热力学过程所描述的,液相α应该是包含了界面相的整个液滴。同时,对液相α采用吉布斯界面热力学基本方程进行定量描述时,式dAα=—pαdVα+μαdnα+σdAS中的pα与气相压强pβ应该相等。本文作者在论文[21]中,从界面热力学基本方程的建立过程以及界面热力学基本方程中压强的数学意义两个角度分别给予了证明。对于整个液滴(含界面相)而言,其吉布斯界面热力学基本方程中的压强为平衡气相压强,所谓的附加压力只是指界面相对体相的。对于第二个热力学过程,既然pα=pβ,dVβ+dVα=0,则有—pαdVα—pβdVβ+σdAS=σdAS≠0。由此可见,对于第二个热力学过程,式(3)并不等于零。
另外,对液相α采用吉布斯界面热力学基本方程进行定量描述时,式dAα=—pαdVα+μαdnα+σdAS中的μα应该为平面液体的化学势。假定式(6)所说的化学势相等是正确的,这也就是说平面液体的化学势与气相化学势相等,而不是式(1)所说的附加压力作用下的体相液体化学势与气相化学势相等。
综合以上对弯曲界面气液两相平衡热力学判据的推导过程分析,由于建立了违背热力学第一定律的、不存在的热力学过程,同时混淆了吉布斯界面热力学基本方程中压强与化学势的取值,本文认为教材[16—20]所提供的证明过程并不能得出弯曲界面气液两相相平衡时化学势相等的热力学判据。
2.2 弯曲界面气液两相相平衡时化学势相等的热力学判据的证明新方法
气液两相化学势相等是可以作为弯曲界面两相平衡的热力学判据的,只是教材[16—20]所提供的证明过程存在问题。
教材[16—20]所提供的证明过程均采用吉布斯界面热力学基本方程,以亥姆霍兹自由能A为例,将A看成四元函数,即A=f(T,V,n,AS)。对于存在弯曲界面的情况,该函数中自变量物质的量n的变化引起其他自变量V、AS的变化。如果要写成式(4)所示的全微分形式,必须使得n、V、AS三个自变量相互独立,为此,吉布斯界面热力学基本方程采取的措施是对式(4)中的偏微分项采用平面相的数值,即式(4)中p、μ和σ均采用平面液体及平面气体的数值,此时化学势的定义为
既然弯曲界面存在时,物质的量n的变化会使得表面积AS变化,则可以不将表面积AS作为自由能函数的自变量,即将描述弯曲界面中气液两相的亥姆霍兹自由能看成三元函数A=f(T,V,n)。将表面相自由能的贡献放入化学势中,即弯曲界面表面积的变化影响化学势的数值,此时化学势的定义为在这种情况下,可以得出弯曲界面中化学势相等的热力学判据,证明过程如下。
仍以图1所示小液滴为研究对象。亥姆霍兹自由能函数只引进三个自变量,即A=f(T,V,n),表面积AS不作为自变量引入。恒温恒容条件下,依据dAβ+dAα=0,有式(7)、式(8)和式(9)。

依据pα=pβ,dVβ+dVα=0,dnα=—dnβ=dn,可得式(10)。

式(10)所提供的弯曲液面两相平衡的相平衡判据也是气液两相化学势相等,但式(10)中的化学势的定义必须采用相对于式(1)所提供的化学势相等的热力学判据,式(10)的证明过程是严谨的。同时,式(10)所提供的化学势相等的热力学判据不需要吉布斯建立化学势相等判据时需要忽略表面相能量的假设前提。
既然气液两相化学势相等可以作为弯曲界面相平衡的热力学判据,如何计算式(10)中的化学势?教材[22]提供了一种计算方法。
对于图1所示纯净小液滴,当有dn mol的物质转移进来时,表面积增大。由于dn mol物质迁移导致的体系总亥姆霍兹自由能增加还包括了表面能的增加,见式(11)。
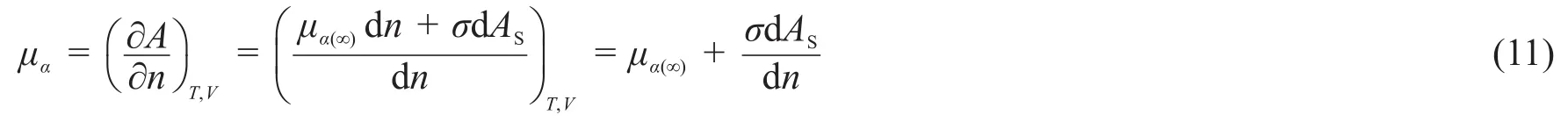
为了更好地区分不同条件下定义的化学势,式(11)中采用教材[22]提供的标注方法,采用μα(∞)表示无限大平面液体的化学势。

将式(12)代入气液两相相平衡判据式(10),可得式(13)。

将化学势的表达式代入式(13),得开尔文方程,即式(14)。

从上述推导过程看,该方法是将整个液滴看成一个整体,式(10)中的μα相当于将表面过剩能σdAS折算进来之后,整个液滴的平均化学势。但是,依据表面过剩能σdAS的含义,σdAS不仅包含了气液界面层中液相部分的过剩能,同时也包括了界面层中气相部分的过剩能,尽管大多数情况下将气相部分的过剩自由能忽略掉。但严格意义上讲,将界面相中气体与液体共有的过剩能量σdAS全部转化为液滴的化学势是不严谨的。所以,式(10)中气相和液相的化学势的计算存在如何定量分配过剩能量σdAS的问题。忽略界面相中气体的过剩自由能,将其全部给予液相只是一个没有办法的办法。
3 一种弯曲界面气液两相相平衡热力学新判据的推导及其应用
3.1 一种弯曲界面气液两相相平衡热力学新判据的推导
采用式(10)提供的化学势相等作为弯曲界面相平衡判据时,对化学势的定量计算严格意义上讲需要对表面过剩能σdAS在界面相中的气相和液相中进行定量分配。本节提供一种无须对σdAS进行分配的弯曲界面两相相平衡新判据。
本节采用吉布斯界面热力学基本方程进行推导。仍以图1所示球状液滴与气体平衡状态为对象进行分析。在恒温恒容条件下,当气相β中有dn mol的物质进入液相α时,依据吉布斯界面热力学基本方程,把气相和液相当成一个整体来考虑。依据dAβ+dAα=0,有式(15)。
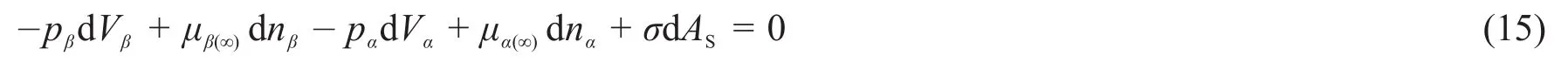
式(15)中的表面过剩能σdAS包含了气相和液相的过剩能。将pα=pβ,dVβ+dVα=0,dnα=—dnβ=dn代入式(15)后有式(16)。

式(16)即弯曲液滴气液两相相平衡热力学判据。式(16)中μα(∞)与μβ(∞)分别为无限大平面液体与无限大平面气体的化学势。
当达到热力学平衡的气体与液体表面均为平面时,气相有dn mol物质进入液相时,气液表面积没有变化,即dAS=0,所以,对于平面液滴,气液两相相平衡的热力学判据为μα=μβ。
式(16)所提供的平衡判据巧妙地避开了表面过剩能σdAS在界面相中气相与液相之间的定量分配,而是把液滴和气体作为整体来考查。该判据的成立要求气液界面张力σ不随液滴半径的变化而变化,也不随气相压强的变化而变化,同时,在对AS进行定量计算时,可近似假定依据液滴半径计算。
3.2 基于新判据的开尔文方程推导
依据式(16)可直接推导出开尔文方程。将式(16)变形得式(17)。

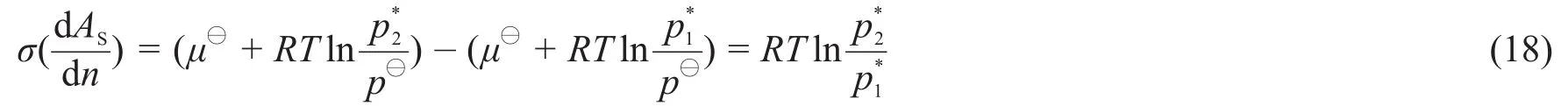

以上推导过程也表明,开尔文方程的成立不需要教材[16—20]所要求的和这两个前提条件。
综上所述,本节提供的弯曲界面气液两相相平衡新判据是依据吉布斯界面热力学基本方程从相平衡时热力学第二定律入手进行推导的,推导过程物理模型清晰,热力学意义明确易懂。
4 结论
1)由于建立了违背热力学第一定律的热力学过程,同时混淆了吉布斯界面热力学基本方程中压强与化学势的取值,现有的关于弯曲界面气液两相相平衡时化学势相等的热力学判据的证明过程是错误的;
2)将描述弯曲界面中气液两相的亥姆霍兹自由能看成三元函数A=f(T,V,n),将界面过剩自由能放入化学势中,弯曲界面两相化学势相等可以作为弯曲界面相平衡的热力学判据。但由于界面过剩能σdAS无法定量分配给界面相中的气相和液相,使得该判据中的化学势计算存在不严谨之处;
3)本文提供了一种无须对表面过剩能σdAS进行定量分配的弯曲界面气液两相相平衡的热力学新判据,即—μ气(∞)dn+μ液(∞)dn+σdAS=0。以该判据进行开尔文方程推导时,开尔文方程的成立不需要和这两个前提条件。
[1]周 鲁.物理化学教程.北京:科学出版社,2002.
[2]杨永华.物理化学.北京:高等教育出版社,2012.
[3]胡 英.物理化学.第5版.北京:高等教育出版社,2007.
[4]邵光杰.物理化学.哈尔滨:哈尔滨工业大学出版社,2002.
[5]傅 鹰.化学热力学导论.北京:科学出版社,1964.
[6]傅献彩,沈文霞,姚天扬.物理化学(下册).第5版.北京:高等教育出版社,2006.
[7]印永嘉,奚正楷,张树永.物理化学简明教程.第4版.北京:高等教育出版社,2007.
[8]Atkins,P.W.;Paula,J.D.Atkins'Physical Chemistry;Oxford University Press:New York,2014.
[9]向井楠宏.高温熔体的界面物理化学.北京:科学出版社,2009.
[10]Berg,J.C.An Introduction to Interfaces of Colloids:the Bridge to Nanoscience;World Scientific Publishing Co.Pte.Ltd.:Singapore, 2009.
[11]顾惕人.表面化学.北京:科学出版社,1994.
[12]李爱昌.大学化学,2013,28(2),81.
[13]吴金添.化学通报,1997,No.7,47.
[14]马延贵.化学世界,1985,No.4,136.
[15]张福田.分子界面化学基础.上海:上海科学技术文献出版社,2006.
[16]王竹溪.热力学.北京:高等教育出版社,1955.
[17]王竹溪.热力学简程.北京:人民教育出版社,1964.
[18]王竹溪.热力学简明教程.北京:商务印书馆,1975.
[19]王竹溪.热力学.第2版.北京:北京大学出版社,2005.
[20]王竹溪.热力学.第2版.北京:北京大学出版社,2014.
[21]章 俊,肖赛君,孔 辉,张博文,唐 鑫.化学通报,2015,78(10),953.
[22]彭笑刚.物理化学讲义.北京:高等教育出版社,2012.
Discussion on Thermodynamic Criterion of Equal Chemical Potential for Phase Equilibrium between Gas and Liquid in Curved Interface
XIAO Sai-Jun*LIU Jian YIN Zhen-Xing ZHANG Jun
(School of Metallurgy Engineering,Anhui University of Technology,Ma'anshan 243002,Anhui Province,P.R.China)
Thermodynamic criterion of equal chemical potential for phase equilibrium between gas and liquid in curved interface has been introduced in many textbooks and papers.In this paper,the process of the derivation of the criterion is proved to be wrong with analysis.In order to solve the problem,a proof procedure for the thermodynamic criterion of equal chemical potential is provided based on the definition of chemical potential.At the same time,a new thermodynamic criterion of two-phase equilibrium in curved interface is established using Gibbs interface thermodynamics and a new method for derivation of Kelvin equation is put forward based on the thermodynamic criterion of two-phase equilibrium.The new criterion which is derived directly from the second law of thermodynamics has a specific thermodynamic significance and clear physical model.
Phase equilibrium between gas and liquid in curved interface;Chemical potential; Kelvin equation
G64;O6
10.3866/PKU.DXHX201603027
*通讯作者,Email:jxddroc@126.com
国家自然科学基金(51404001);安徽省留学回国人员创新创业扶持计划资助项目[2016]
www.dxhx.pku.edu.cn
