Biodiversity and phylogenetic analysis of the gut microbiome of Euphausia superba Dana from the Rose Sea of the Antarctic Ocean
2016-02-01YUANLidongLILingzhiTIANXiaoqingTANGYingyingFANChengqiHUANGHongliangYANGQiao
YUAN Lidong, LI Lingzhi, TIAN Xiaoqing, TANG Yingying,FAN Chengqi, HUANG Hongliang & YANG Qiao,3,4*
1 Key Laboratory of East China Sea & Oceanic Fishery Resources Exploitation & Utilization, Ministry of Agriculture,East China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Shanghai 200090, China;
2 Shanghai Ocean University, Shanghai 201306, China;
3 State Oceanic Administration Key Laboratory for Polar Science, Polar Research Institute of China, Shanghai 200136,China;
4 Department of Microbiology, Oregon State University, Corvallis 97333, OR, USA
1 Introduction
Natural products derived from marine microorganisms have become key sources for discovering novel marine drugs,which can be used to treat major diseases such as cancer and cardiovascular ailments[1]. Symbiotic bacteria and their hosts have engaged in long-term, co-evolutionary relationships,and these bacteria are influenced by the genetics, lifestyle,and environment of their hosts. Moreover, symbiotic bacteria and their hosts exchange important genetic materials via horizontal gene transfer, as well as a variety of metabolites.Thus, natural metabolites from the symbiotic bacteria of marine organsims have become a new focus for the discovery of novel lead compounds and innovative drugs[2].
Polar microorganisms have their own unique, adaptive survival mechanisms to adapt to their special natural habitats,and they are rich resources of novel microbial species, genes,and natural bioactive products. Euphausia superba Dana is an Antarctic krill, which comprise a group of shrimp-like crustaceans that occupy a unique position in the Antarctic Ocean ecosystem[3–7]. Its unique lifestyle makes it a natural trap for Antarctic extremophilic microorganisms, which include a large number of symbiotic intestinal microbes[8–9].Studies have shown that the vital life processes of a host,including nutrient metabolism, evolutionary development,immunity, disease resistance, and environmental adaptation,are affected by its intestinal microbiome[10]. A typical microbiome genome contains over 100-fold more genes than that of its host genome, and it has unique features that are not available in its host[11–13].
Currently, no study has examined the biodiversity of the symbiotic intestinal microbiome of Antarctic krill.Here, we used high-throughput sequencing to analyze the species biodiversity of the gut microbiome of E. superba.Additionally, we isolated cultivable strains using the pure culture technique, and we performed a phylogenetic analysis of the 16S rDNA sequences. The results will provide useful scientific clues for understanding the ecological relationship between the intestinal microbiome and Antarctic krill, and they may lead to the discovery of new marine symbiotic bacteria, which could be a useful resource for novel marine drug discovery.
2 Materials and methods
2.1 Materials
Sample of E. superba Dana were obtained during the 29th Chinese National Antarctic Expedition in 2012/2013. The sampling position was 169°33′E and 74°26′S in the Rose Sea. The krills were caught with a specific midwater frame trawl[14]. Immediately after being caught, the krills were incubated at 4°C in sterile plastic vessels on board, or they were frozen and maintained at −80°C in 25% (v/v) glycerol.Zobell marine media 2216E, ISP1, ISP2, M1, M2,M4, M5, and HP were used for microbial strain isolation and cultivation. Natural seawater from the Antarctic Ocean was used to prepare the media.
2.2 DNA extraction and polymerase chain reaction(PCR) amplification
Genomic DNA of the sample was extracted using the Wizard® DNA Kit (Promega, Madison, WI, USA)according to the manufacturer’s instructions. The quality of the extracted DNA was checked by 0.8% agarose gel electrophoresis and by spectrophotometrically measuring the 260 nm/280 nm ratio. The V3-V4 region of bacterial 16S rRNA was amplified by PCR for high-throughput pyrosequencing. The bacterial 16S rRNA gene V3-V4 region was amplified using the universal forward primer 515F (5′–GTGCCAGCMGCCGCGG–3′) and the reverse primer 907R (5′–CCGTCAATTCMTTTRAGTTT–3′).PCR amplifications of the 16S rRNA V3-V4 region was performed as follows: an intial denaturation at 95°C for 3 min, followed by 27 cycles of 95°C for 30 s, 55°C for 30 s,and 72°C for 45 s, with a final extension of 72°C for 10 min.The amplicon mixture was applied to the HiSeq 2500 MiSeq Genome Sequencer (Illumina, San Diego, CA, USA).
2.3 High-throughput sequencing
Extraction of high-quality DNA was performed, and raw sequences were selected, while low-quality sequences were removed. Alignment of the high-quality sequences was performed using PyNAST and UCLUST. The unique sequence set was classified into operational taxonomic units(OTUs) at a 97% identity threshold using UCLUST. Chimera Slayer was applied to remove potential chimeric sequences from the representative set of OTUs. Alpha diversity was used to analyze the microbial diversity. The indices for community richness included the Chao1 estimator (http://www.mothur.org/wiki/Chao) and the abundance-based coverage estimator (Ace) (http://www.mothur.org/wiki/Ace).The indices for community diversity included the Shannon index (http://www.mothur.org/wiki/Shannon) and the Simpson index (http://www.mothur.org/wiki/Simpson). The sequencing depth index used Good’s coverage (http://www.mothur.org/wiki/Coverage). Mothur (version v.1.30.1, http://www.mothur.org/wiki/Schloss_SOP#Alpha_diversity) was used for data analysis[16].
2.4 Isolation of cultivable strains
Cultivable bacteria were isolated and maintained in Zobell 2216E media that was prepared with aged and filtered natural seawater. Bacterial isolation was performed by harvesting 1 mL of a gut sample. Following centrifugation, the spent medium was removed, and the cell pellet was re-suspended in 100 μL of sterile seawater by vortexing. A 10-fold dilution series of the cell suspension was created, and 100 μL of each dilution was spread onto 2216E agar plates and incubated in dark at 15°C for 2 weeks. Bacterial colonies with distinct colony morphologies were picked. Bacterial isolates were grown in 2216E broth, and the cells were stored at −80°C in 25% (v/v) glycerol.
2.5 Phylogenetic analysis
Bacterial genomic DNA was extracted from a 1-mL culture of exponentially growing bacteria using the Wizard DNA Extraction Kit according to the manufacturer’s instructions.PCR of the 16S rDNA was performed using the universal primers 27F (5′–AGAGTTTGATCCTGGCTAG–3′) and 1492R (5′–GGTTACCTTGTTACGACTT–3′)[17]. The PCR volume was 50 μL, and the reactions were performed as follows: an initial denaturation at 94°C for 5 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 1 min, followed by a final extension at 72°C for 10 min. PCR products were analyzed using 1% agarose gel electrophoresis and sequenced, and the similarity of 16S rRNA gene sequences was analyzed by a Basic Local Alignment Search Tool search of the GenBank database at the National Center for Biotechnology Information. A phylogenetic tree was constructed by the neighbor-joining method using MEGA software (version 2.0)[18].
3 Results
3.1 Quality control data for the high-throughput sequencing sample
The species diversity of the symbiotic intestinal microbiome was determined by high-throughput sequencing. The quality of the sequencing sample was analyzed, and the results are shown in Table 1. Trimmed sequences with lengths of 301–400 bp accounted for more than 99.98% of the total sequences, indicating that the sample data were suitable for the subsequent high-throughput sequencing analysis.
3.2 Biodiversity index analysis
An analysis of the dilution curve of the sample was performed using the random sequence sampling method.A rarefaction curve was constructed using the number of sequences and the representative number of OTUs (Figure 1). The results showed that there was sufficient depth for the high-throughput sequencing of the sample. The diversity results, including microbiome abundance (community richness), the Chao and Ace indices, bacterial diversity(community diversity), the Shannon and Simpson indices,and the sequencing depth index (coverage), are shown in Table 2. The Shannon–Wiener curve of the sequencing sample is shown in Figure 1. These results illustrate the high species biodiversity of the symbiotic microbiome.values greater than 97%, the OTU abundance, and the number of sequences, as determined by an OTU clustering analysis of the high-throughput sequencing sample.
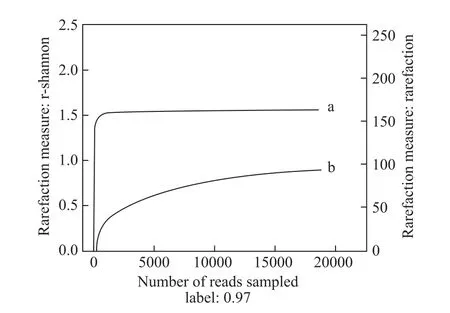
Figure 1 The Shannon–Wiener (a) and rarefaction curves (b) of the high-throughput sequencing sample.
3.4 Taxonomic analysis
The Ribosomal Database Project (RDP) classifier of a Bayesian algorithm was used to statistically analyze the microbial community composition, and it was performed using representative sequences of OTUs with similarity values greater than 97%. This analysis was performed at each classification level. The results showed that 61 known species matched GenBank database records at the species level (Figure 2). In addition, approximately 7% of the species were previously unknown. The community contained 56 genera (Figure 3), eight of which, including Arthrobacter,Bacillus, Candidatus, Lactococcus, Lysinibacillus,Leuconostoc, Solibacillus, and Vibrio, were dominant, as was the family Vibrionaceae. The results illustrate the richness of the symbiotic microbial community in the E. superba gut.

Table 1 Quality control data for the high-throughput sequencing sample

Table 2 Community richness and diversity data of the highthroughput sequencing sample
3.3 OTU distribution statistics
OTUs are defined operational classification units in phylogenetic or human population genetics research, and they are used for comparative analyses. Table 3 shows the representative OTUs of the sample with sequence similarity
3.5 Biodiversity analysis of the cultivable strains
We used a variety of selective isolation media to isolate cultivable symbiotic strains from the gut microbiome of E. superba. Then, their 16S rRNA genes were sequenced and aligned with sequences in the National Center for Biotechnology Information database (Figure 4). Eightyone cultivable strains were obtained. A phylogenetic analysis showed that they belonged to 36 species,including Proteobacteria, Actinobacteria, Firmicutes and Bacteroides spp. Among them, Proteobacteria spp.,particularly α-Proteobacteria and γ-Proteobacteria spp.,were dominant, accounting for approximately 68% of the cultivable strains. These strains belonged to 36 genera, of which Albirhodobacter, Pseudoalteromonas, Arthrobacter,and Bacillus were dominant. The 20 isolated actinomycetes strains belonged to 10 genera, including Streptomyces,Isoptericola, Mobilicoccus, Leifsonia, Arthrobacter,Rhodococcus, Brevibacterium, Salinibacterium, Subtercola,and Chryseoglobus.

Table 3 OTUs and sequences numbers of the high-throughput sequencing sample
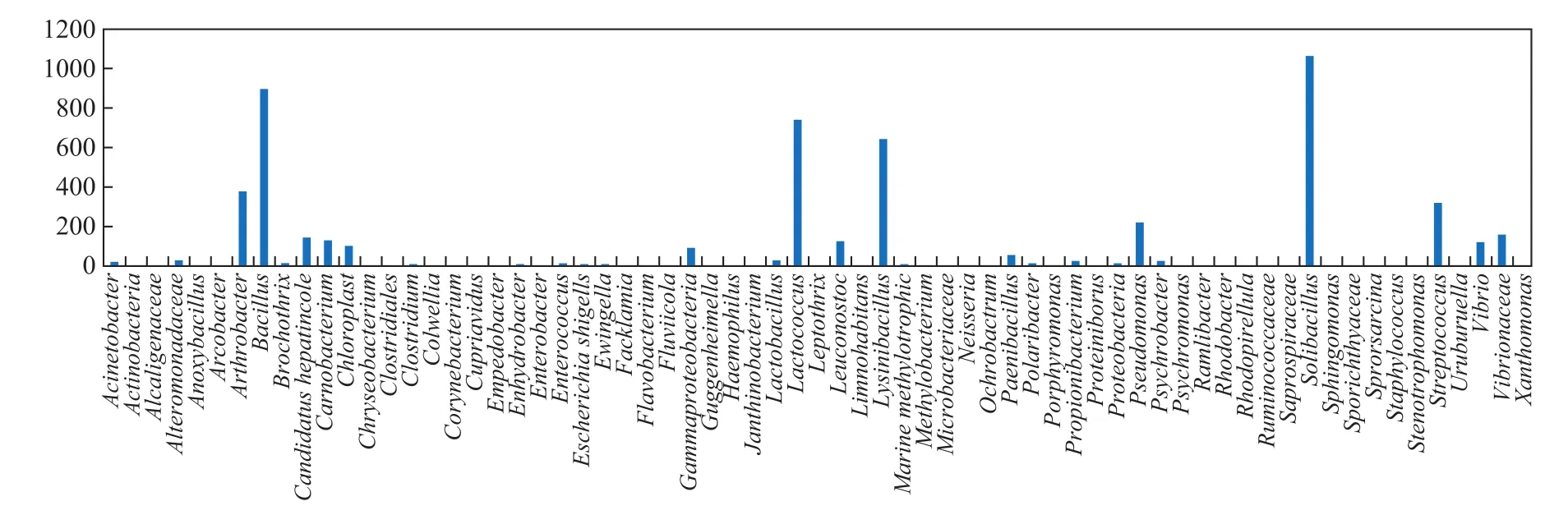
Figure 2 Species biodiversity and abundance of the gut microbiome of E. superba.
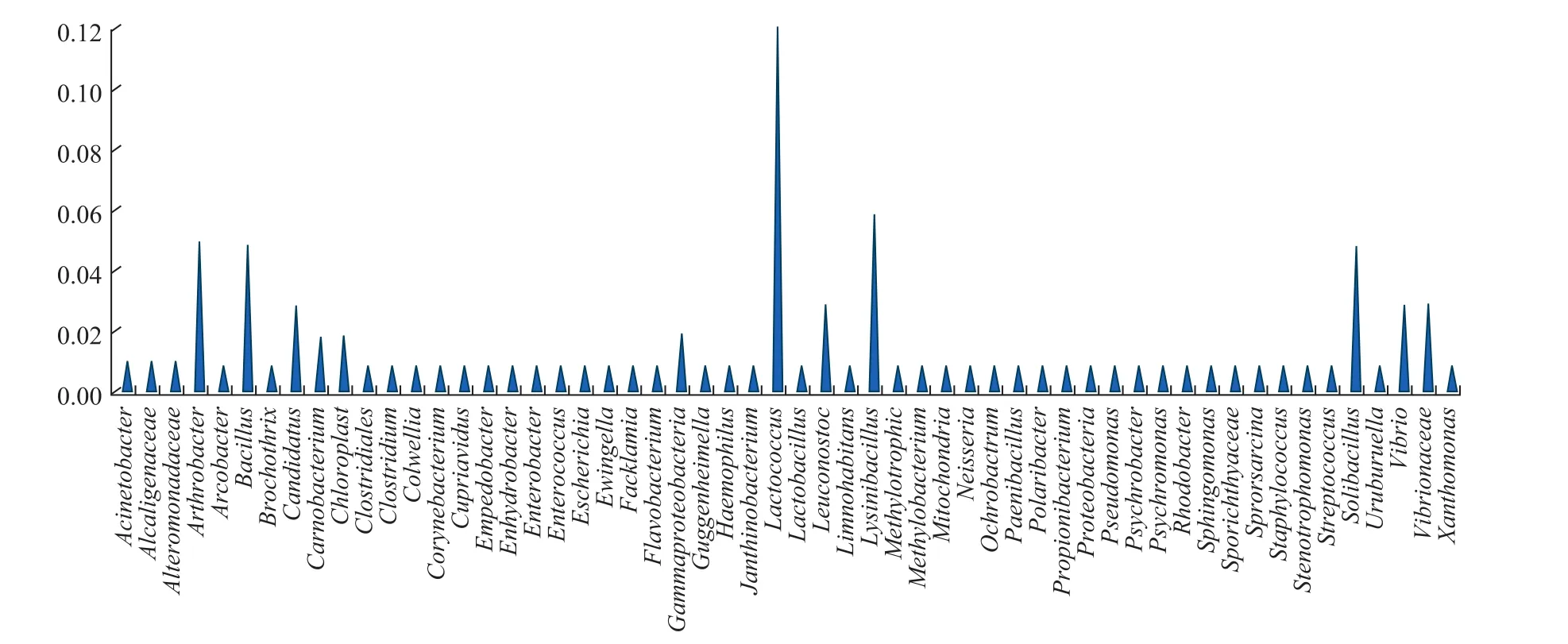
Figure 3 Microbial distribution at the genus level of the gut microbiome of E. superba.
4 Discussion
Polar microorganisms live in an extreme natural environment. Thus, because of natural selection, they have evolved unique mechanisms enabling them to adapt to this extreme environment. As a result, they have great potential to generate new, biologically active natural products[19–23].Antarctic krill are key species in the Southern Ocean ecosystem. Their symbiotic intestinal microbiomes have been proven to be new sources of symbiotic bacteria and their associated metabolites[9,24–26]. In the present study,culture-independent, high-throughput sequencing was used to reveal the species diversity of the symbiotic intestinal microbiome of E. superba. Fifty-six genera were found in the microbiome, and they contained a large number of unknown and uncultivable strains, which indicates that there is a high probability of finding new marine microbial species in this special habitat[8,9,25]. Additionally, 81 cultured strains were isolated by the pure culture method. The phylogenetic analysis indicates that the microbiome of E.superba comprises 36 genera, in which those belonging to the Proteobacteria were the most abundant, followed by those belonging to the Actinobacteria. The dominant genera included Albirhodobacter, Pseudoalteromonas, and Arthrobacter, as well as psychrotrophic spp. The rare genera included Isoptericola, Mobilicoccus, Leifsonia, Arthrobacter,Rhodococcus, Brevibacterium, Salinibacterium, Subtercola,and Chryseoglobus.
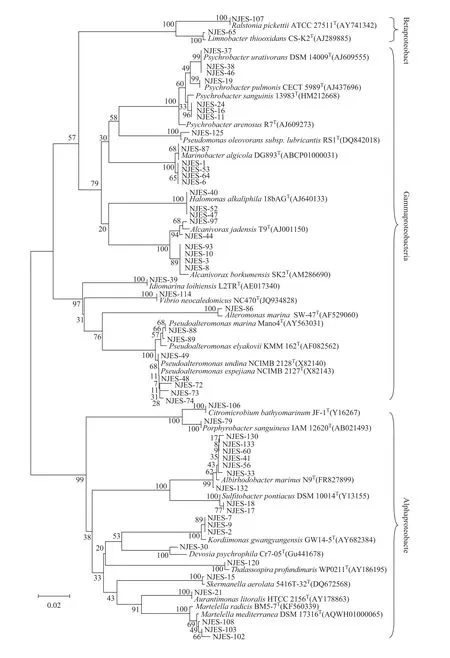
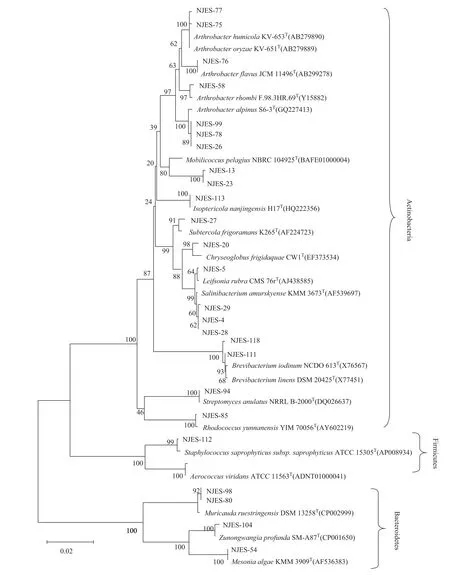
Figure 4 Phylogenetic tree of the cultivable bacteria of the gut microbiome of E. superba based on 16S rRNA sequences.
The species diversity of the symbiotic intestinal microbiome that was revealed in this study may provide useful scientific clues for optimizing strategies to culture and isolate such species, which will enable researchers to track bioactive metabolites and screen for novel bioactive natural products. Our results also showed that the number of cultivable symbiotic strains that were isolated by the pure culture method was much lower than the species abundance revealed by culture-independent high-throughput sequencing[24–26]. This indicates that better culture strategies are needed to improve the cultivability of microbial species,especially marine symbiotic microorganisms in unique habitats. Developing such culture strategies will reveal the true picture of the biodiversity of the intestinal microbiome of Antarctic krill[27]. Meanwhile, further research on the isolation of cultivable symbiotic strains of the intestinal microbiomes of Antarctic krill, as well as their potential ecological functions,will receive increasing attention in the related research fields in the near future.
1 Newman D J, Cragg G M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod, 2012, 75(3): 311–335
2 Clay K. Defensive symbiosis: a microbial perspective. Funct Ecol,2014, 28(2): 293–298
3 Puglisi M P, Sneed J M, Sharp K H, et al. Marine chemical ecology in benthic environments. Nat Prod Rep, 2014, 31(11): 1510–1553
4 Brierley A S, Fernandes P G, Brandon M A, et al. Antarctic krill under sea ice: elevated abundance in a narrow band just south of ice edge.Science, 2002, 295(5561): 1890–1892
5 Sun S. Antarctic krill. World Sci-tech R D, 2002, 24(4): 57–60
6 Sun S, Liu Y Q. Antarctic Krill and southern ocean ecosystem. Chin J Nat, 2009, 31(2): 88–90, 104 (in Chinese)
7 Liu Y Q, Sun S, Zhang Y S, et al. Distribution and demography of Euphausia superba Dana in Prydz bay during Junuary 2002. Chin J Polar Res, 2011, 23(4): 275–282 (in Chinese)
8 Wang C, Tian X Q, Yang Q, et al. Diversity of secondary metabolites from two Antarctic microbes Rhodococcus sp. NJ-008 and Pseudomonas sp. NJ-011. Open J Mar Sci, 2014, 4(3): 214–220
9 Yang Q. Taxonomic identification and bioactivity screening of the symbiotic bacteria strains of Euphausia superba from Antarctic Ocean. Appl Mech Mater, 2013, 295–298: 173–177
10 Le Chatelier E, Nielsen T, Qin J J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature, 2013,500(7464): 541–546
11 Gomez A, Petrzelkova K J, Burns M B, et al. Gut Microbiome of coexisting Baaka pygmies and bantu reぼects gradients of traditional subsistence patterns. Cell Rep, 2016, 14(9): 2142–2153
12 Gill S R, Pop M, DeBoy R T, et al. Metagenomic analysis of the human distal gut microbiome. Science, 2006, 312(5778): 1355–1359
13 Eckburg P B, Bik E M, Bernstein C N, et al. Diversity of the human intestinal microbial ぼora. Science, 2005, 308(5278): 1635–1638
14 Li L Z, Huang H L, Qu T C, et al. A correlation study between the marine environment and the spatial-temporal distribution of Antarctic krill (Euphausia superba Dana) in Prydz Bay. J Fish Sci China, 2015,22(3): 488–500 (in Chinese)
15 Nacke H, Thürmer A, Wollherr A, et al. Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS One,2011, 6(2): e17000
16 Andersson A F, Lindberg M, Jakobsson H, et al. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One,2008, 3(7): e2836
17 Lane D J. 16S/23S rRNA sequencing//Stackebrandt E, Goodfellow M.Nucleic acid techniques in bacterial systematics. New York: Wiley,1991: 115–175
18 Akbar A, Zhang L, Zhu L F, et al. Sphingomonas hylomeconis sp.nov., isolated from the stem of Hylomecon japonica. Int J Syst Evol Microbiol, 2015, 65(11): 4025–4031
19 Liu Y, Zhang F, Ling Y, et al. Diversity and community composition of bacterioplankton in the Bering Sea during summer 2010. Chin J Polar Res, 2013, 25(3): 113–123 (in Chinese)
20 Yuan M, Yu Y, Li H Y, et al. Molecular identification, antimicrobial activity and fermentation conditions of antibiotic substances of marine actinobacteria Nocardiopsis sp. Y18. Chin J Polar Res, 2014, 26(3):292–298 (in Chinese)
21 Zeng Y X, Chen B, Zou Y, et al. Polar microorganisms, a potential source for new natural medicines-a review. Acta Microbiol Sin, 2008,48(5): 695–700 (in Chinese)
22 Bredholdt H, Galatenko O A, Engelhardt K, et al. Rare actinomycete bacteria from the shallow water sediments of Trondheim fjord,Norway: isolation, diversity and biological activity. Environ Microbiol, 2007, 9(11): 2756–2764
23 Yu Y, Li H R, Zeng Y X, et al. Isolation and phylogenetic assignation of actinomycetes in the marine sediments from the Arctic Ocean. Acta Oceanol Sin, 2005, 24(6): 135–142
24 Zhang X L, Yang Q, Fan C Q, et al. Isolation, identification and screening of nitric oxide targeted inhibition bioactivity of a symbiotic/epiphytic bacterium from Euphausia superba. Mar Fish, 2011, 33(4):436–441 (in Chinese)
25 Zhang X L, Lu Y N, Fan C Q, et al. Isolation, identification and bioactivity analysis of a new PQQ-producing marine bacteria from the Antarctic ocean. Mar Fish, 2012, 34(1): 89–95 (in Chinese)
26 Wang C, Yang Q, Ma LY, et al. Secondary metabolites of Pseudomonas sp. NJ-013 from the Antarctic. Mar Fish, 2014, 36(4):350–356 (in Chinese)
27 Rappé M S, Connon S A, Vergin K L, et al. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature, 2002,418(6898): 630–633
杂志排行

Advances in Polar Science的其它文章
- Norwegian contributions to Arctic environmental sciences from the 1880s to the third International Polar Year
- Meteorite classification for building the Chinese Antarctic Meteorite Depository—Introduction of the classification of 500 Grove Mountains meteorites
- Structure and seasonal variability of fronts in the Southeast Indian Ocean along sections from Fremantle, Australia to Antarctic Zhongshan Station
- Outreach channels for polar science: an expedition to Kerguelen Islands as a case study
- Potential methane production rates and its carbon isotopic composition from ornithogenic tundra soils in coastal Antarctic
- Expression analysis of heat-shock protein gene Hsp845 in the Antarctic psychrotrophic bacterium Psychrobacter sp. G under temperature and salinity stress