分子动力学模拟及在生物大分子模拟领域的应用
2016-01-31刘冠辰
刘冠辰
(吉林化工学院 材料科学与工程学院,吉林 吉林 132022)
分子动力学模拟及在生物大分子模拟领域的应用
刘冠辰
(吉林化工学院 材料科学与工程学院,吉林 吉林 132022)
摘要:简要介绍了分子动力学的发展历史、基本理论、基本步骤以及其作为基本研究手段来进行生物大分子模拟领域的应用.
关键词:分子动力学;生物大分子
分子动力学(Molecular Dynamics,MD)模拟是指研究在一定的条件下微观的粒子体系中,建立粒子运动方程并通过求解粒子运动方程来得到粒子相空间的运动状态来得到微观粒子的运动规律,并通过微观粒子的运动规律进一步得到宏观体系基本规律和特性.
1分子动力学(MD)模拟发展
早在1957年,Alder[1]等人初次运用分子动力学模拟方法来计算凝聚态下的气体和液体状态方程,开创了把分子动力学模拟方法应用在研究物质宏观性质的先例.1967年,verlet提出逐步去计算对粒子的速度、加速度以及距离的方法,即著名的verlet算法.1972年,Less[2]等人将分子动力学模拟方法运用到具有速度梯度的非平衡系统.1980年,Andersen[3]等人提出了恒定压强分子动力学模拟方法.1981年,parrinello[4]等人将恒定压强分子动力学模拟方法扩展到元胞形状发生改变的范围.1983年,Gillan[5]等人又将分子动力学模拟方法推广到具有温度梯度的非平衡系统.1984年,Nosé[6]等人又提出了恒定温度下分子动力学模拟方法.1985年,Car[7]等人运用分子动力学与密度泛函理论相结合的第一性原理分子动力学方法来解决半导体和金属等势函数模型化困难问题.1991年,Cagin[8]等人又将分子动力学模拟方法去处理巨正则系统的吸附问题.2007年,Bussi[9]等人在Nosé方法上进行了改进,增加了随机项来使动能分布更为准确.
近几年,随着计算机计算速度的提升和分子模拟理论的快速发展,分子动力学(Molecular Dynamics,MD)模拟作为分子模拟方法其中的一种,在化学、生物学、材料学等多个自然科学领域得到了广泛的应用.
2分子动力学
2.1 分子动力力学基本理论
分子动力学模拟方法是利用经典牛顿力学来进行大分子体系运动的模拟,这和量子化学计算方法不同,量子化学是用薛定谔方程为指导依据的,分子动力学则是以牛顿运动方程为理论基础.
分子动力力学基本理论如下:在分析的整个系统中,假设含有n个分子或者构成分子的原子,此时这个系统的总能量为系统内部分子的总势能和总动能之和,其中分子或者原子的位置函数为总势能V,这包括分子间的相互作用的范德华力Vvdw和分子内部势能Vint
即V=Vvdw+Vint
(1)
一般分子间相互作用的范德华力可以用构成分子的各个原子对之间的范德华力相互叠加来表示,即为:
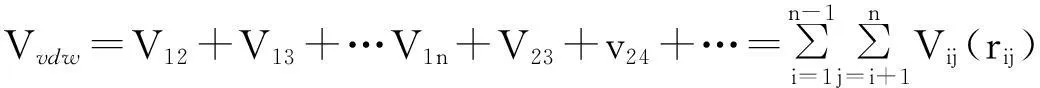
(2)
其中:rij表示为原子i和原子j之间的距离;用原子之间的各类型内坐标(如键伸缩、二面角扭转..)的总势能来表示分子内部势能Vint
由经典力学可知,在整个模拟系统中原子i所受的力为势能的梯度:
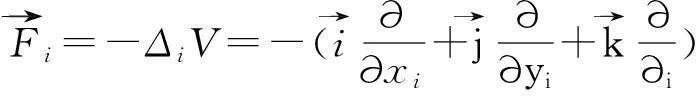
(3)
据此,通过牛顿运动方程可以得到i原子的加速度为:
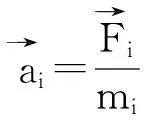
(4)
将上式对时间积分,人们可以预测i原子经过时间t后的速度和相应的位置.与表示粒子的位置与速度,上标0表示各个物理量的初始值.
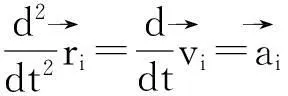
(5)
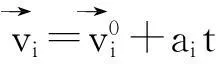
(6)

(7)
由以上公式推导可知,分子动力学基本理论是以牛顿运动定律为理论基础的.首先,通过确定系统中各个原子的位置来计算总势能V,公式(3)计算出各个原子受到范德华力,公式(4)计算出各个原子受到的加速度,在公式(7)中,假设t=δt(δt 来表示时间间隔极短),进一步可以得到在经过δt后的各个原子的速度和位置,多次重复这个过程步骤,并由新的原子位置来计算整个体系势能、各个原子受到范德华力以及各个原子受到的加速度,进而计算经过δt后的各个原子的速度和位置,多次循环后得到在不同时刻时各个分子或者构成分子的原子的位置、速度以及加速度;我们一般将这种所有时刻上的分子位置为运动轨迹(trajectory).
又由于这个体系中的分子或构成分子的原子之间的相互作用比较复杂,不能用一般的解析法来计算分子的运动方程,只能用差分法来近似计算分子的运动方程.目前常用的一些差分算法有Gear算法[10]和Verlet算法[11],其中在分子动力学中,以Verlet提出的Verlet算法来计算分子运动方程的方法比较普遍,Verlet算法有进一步进行了改进为更有效的类Verlet算法(如蛙跳法(leap-frog)[12]、velocity-Verlet算法[13]).
2.2 分子动力学模拟研究基本步骤
分子动力学最重要的特性之一就是可以观察体系随时间演变的过程,随着计算机水平的提升,分子动力学目前已经可以达到毫秒级(ms)的时间尺度;分子动力学模拟的步骤主要有:
第一步为确定初始构型.通常选取较低能量的起始构型,这样更接近分子模拟系统.在生物大分子体中,一般先用分子力学方法进行优化,获的较低能量的初始构型,然后在进行分子动力学模拟.而对于小分子的构象来说,可以通过量子力学或者分子力学方法来求得.在起始构型确定后,构成分子的各个原子要赋予一定速度,这个速度将由玻尔兹曼分布随机生成,在这个阶段,由于各个原子速度分布符合玻尔兹曼统计,整个体系处于恒定的温度;另外,要进一步调整随机生成的各个原子运动速度,让整个体系在各个方向上的动量之和为零来确保这个体系没有平动位移.
第二步为平衡相(equilibration).由于第一步中模拟体系已得到确定,这一步中模拟体系将进入平衡相;而在构建平衡相的时候需要对体系的温度、构象等参数加以控制和调整,进而确定整个体系是否达到平衡.
第三步为生产相(productionrun).当模拟体系在经过平衡相过程之后,下一步将进入到生产相过程.此时模拟体系中的分子或者分子中的原子开始进行相互作用力,如发生吸引或排斥,并开始进行初始速度运动,并根据预先给定的粒子间相互作用势和牛顿力学来计算各个粒子的运动轨迹,进而抽取出相关的数据和样本来用于分析计算.
第四步为进一步分析处理计算结果.
3分子动力学模拟在生物大分子领域中的应用
目前,分子动力学模拟已经成为研究生物大分子常用的比较热点的有力工具之一,和其他模拟方法来比,它有着扎实、正确理论基础来支撑并能确保其模拟结果的准确性和适宜性.利用分子动力学模拟可以对生物大分子进行模拟其空间构象、模拟其运动以及进行相关热力学数据计算,并对其实验结果进行预测和指导.分子动力学模拟研究生物大分子在内容上体现在如下:首先,利用计算机对生物大分子进行模拟,来研究这个大分子体系在时间维度上的变化规律.其次,分析生物大分子体系平衡态的性质,根据大分子的热力学数据来进行相关数据计算.最后,进行生物大分子空间构象的研究.
3.1 在蛋白质上研究的应用
在生物体中最复杂的大分子之一为蛋白质,它是组成人体一切细胞、组织的重要成分.在蛋白质中,某些氨基酸残基还可以被翻译后修饰而发生化学结构的变化来对蛋白质进行激活或调控[14].多个蛋白质可以一起,折叠或螺旋构成一定的空间结构来发挥正常的生物功能.如果蛋白质的折叠或螺旋构象出现轻微错误就会导致构象病的产生,如亨廷顿氏病、帕金森氏病、葛雷克氏病以及阿尔察默病等严重的神经性疾病.1998年,Yong和Peter[15]第一次用分子动力学模拟了微秒级的蛋白质大分子折叠构象,这对蛋白质结构预测、蛋白质复合化研究迈出了第一步,也是蛋白质大分子分子动力学研究的里程碑.随后周耀旗[16-21]也进行了蛋白质折叠的机理以及其热力学性质的研究,Dokholyan和Shakhnovich等[22-23]对蛋白质的折叠核、动力学以及热力学性质进行了更深入一步的研究.因此,研究蛋白质的折叠或螺旋构象以及动态变化对合成特殊功能蛋白质大分子和新功能药物的设计合成有着重要指导意义,这也逐渐成为现代自然科学领域研究的热点之一.
3.2 在核酸模拟上的应用
核酸同蛋白质一样,也是生物大分子,同时在蛋白质的复制和合成中起着储存和传递遗传信息的作用[24].核酸不仅是基本的遗传物质,而且在蛋白质的生物合成上也占重要位置.现已发现近2000种遗传性疾病都和核酸有关,如人类镰刀形红血细胞贫血症是由于患者的血红蛋白分子中一个氨基酸的遗传密码发生了改变,白化病患者则是DNA分子上缺乏产生促黑色素生成的酪氨酸酶的基因所致.肿瘤的发生、病毒的感染、射线对机体的作用等都与核酸有关.而分子动力学模拟在核酸模拟上的研究应用主要集中在蛋白质、细胞结合水、药物大分子等对核酸结构上的影响.如美国北卡罗莱纳大学Singh[25]等人主要采用分子动力学模拟手段来研究核酸链与脱氧核糖核酸之间的相互作用,其研究结果表明核酸不适合生成较适宜的膜结构,这也为今后在药物开发、分子传感器等生物领域的应用有着重要的指导意义.
3.3 在生物药物大分子设计上的应用
分子动力学模拟技术不仅用于蛋白质或核酸之间的相互模拟分析,也可以用于生物药物大分子与蛋白质的相互结合模拟研究,进而可以得到生物药物大分子的蛋白构象来对生物药物大分子进行分子设计.计算机辅助生物药物大分子设计[26-27]可分为直接生物药物大分子设计和间接生物药物大分子设计.直接生物药物大分子设计是指在已知受体结构上来进行生物药物大分子设计的一种方法,而间接生物药物大分子设计是指用已知活性的生物药物大分子分子群对其结构和活性进分析并提出来结构与活性之间的相关性而得到的有效药效基团.如果靶点/靶点配体的结构已知,可以用直接生物药物大分子设计方法来进行新药结构的设计;反之,则用间接生物药物大分子设计的方法.在直接生物药物大分子设计或者是在间接生物药物大分子设计中,生物信息学为我们提供了各种生物大分子结构和序列的信息,我们也利用这些信息开发并建立了可以用于分子模拟计算用的数据库[28-30]如EMBL、SWISSPROT、PDB、GENBANK、BLOCKS、 PIR等以及各种算法软件[31-36]如Apex-3D、GCG、LUDI、DOCK、Catalyst 等,这些方法和手段为计算机辅助生物药物大分子设计模拟提供了先进的科学技术手段,为今后医药生物药物大分子设计开发具有难以估量的意义.
4展望未来
随着科学的不断进步以及计算机软硬件的快速发展,在即将不就的将来分子动力学模拟技术一定会在生物大分子研究领域占主导地位.因为分子动力学模拟技术可以很好的模拟生物大分子中蛋白质、核酸以及生物药物大分子开发的结构、变化的动态过程,将时间尺度推到微秒级别甚至毫秒级别,这种模拟后得到的计算结果与真正实验数据相比更接近,也使得目前在实验室无法完成和实现的研究得以顺利的进行,为此方向填补一项空白.但是在另一个角度说,尽管分子动力学模拟技术发展的比较快速,但是生物体的体系内部结构要比想象中的复杂,而却不确定因素会随着时间变化而变化,这就使得此项技术对生物大分子模拟过程还十分有限,这就要求我们尽可能提高计算软硬件的不断完善,来满足我们解决实际问题的需要;也正是这点要求需要我们在模拟生物大分子研究的同时还需要进行相关的实验,做到将理论与实践相结合,模拟计算指导实验方向,实验结果优化模拟数据,相互揉和才能有效解决生物大分子领域的难题.
参考文献:
[1]BJ Alder,TE Wainwright.Phase Transition for a Hard Sphere System[J].J.Chem.Phys.,1957(27):1208-1219.
[2]AW Less,SE Edwards.The Computer Study of Transport Process under Extreme Conditions[J].J.Phys.C:Solid State Phys.,1975(5):1921-1929.
[3]HC Anderson.Molecular Dynamics Simulations at Constant Press and/or Temperature[J].J.Chem.Phys.,1980(72):2384.
[4]M Parrinello,A Rahman.Polymorphic Transitions in Single Crystals:A New Molecular Dynamics Method[J].J.Appl.Phys.,1981(52):7182.
[5]MJ Gillian.M Dixon.The Calculation of Thermal Conductivities by Perturbed Molecular Dynamics Simulation[J].J.Phys.C:Solid State Phys.,1983(15):869-878.
[6]S Nose.A Unified figuretion of the Constant Temperature Molecular Dynamics Methods[J].J.Chem.Phys.,1984 (81):511.
[7]R Car,M Parrinello.Unified Approach for Molecular Dynamics and Density-Functional Theory[J].Physical Review Letters,1985(55):2471-2474.
[8]T Cargin,BM Pettitt.Molecular Dynamics with a Variable Numbers of Molecules[J].Molecular Physics,1991(72):169-175.
[9]G Bussi,D Donadio,M Parrinello.Canonical Sampling through Velocity Rescaling[J].J.Chem.Phys.,2007(126):014101.
[10] L Verlet.Computer “experiments” on Classical Fluids[J].Phys.Rev.,1967(159):98-103.
[11] 文玉华,朱如曾,等.分子动力学模拟的主要技术[J].力学进展,2003(33):65-73.
[12] RW Hockney,Methods in Computational Physics[M].1970,Academic Press:New York.
[13] WC Swope,HC Andersen,PH Berens,et al.A Computer-Simulation Method for the Calculation of Equilibrium-Constants for the Formation of Physical Clusters of Molecules-Application to Small Water Clusters[J].J.Chem.Phys.,1982(76):637-649.
[14] 张民,郭志红,周鸿立.多糖类物质脱蛋白方法的概况[J].吉林化工学院学报.2013,30(5):44-46
[15] Y Duan,PA Kollman.Pathways to a Protein Folding Intermediate Observed in a 1-Microsecond Simulation in Aqueous Solution[J].Science,1998(282):740-744.
[16] YQ Zhou,M Karplus.Interpreting the Folding Kinetics of Helical Proteins[J].Nature,1999(401):400-403.
[17] A Linhananta,YQ Zhou.The Role of Sidechain Packing and Native Contact Interactions in Folding:Discontinuous Molecular Dynamics Folding Simulations of an All-atom Gō Model of Fragment B of Staphylococcal Protein A[J].J.Chem.Phys.,2002(117):8983-8995.
[18] YQ Zhou,A Linhananta.Role of Hydrophilic and Hydrophobic Contacts in Folding of the Second beta-Hairpin Fragment of Protein G:Molecular Dynamics Simulation Studies of an All-atom Model[J].Proteins:Structure,Function,and Bioinformatics,2002(47):154-162.
[19] YQ Zhou,M Karplus.Folding of a Model Three-helix Bundle Protein:A Thermodynamic and Kinetic Analysis[J].Journal of Molecular Biology,1999(293):917-951.
[20] YQ Zhou,A Linhananta.Thermodynamics of an All-atom Off-lattice Model of the Fragment B of Staphylococcal Protein A:Implication for the Origin of the Cooperativity of Protein Folding[J].Journal of Physical Chemistry B,2002(106):1481-1485.
[21] H Jang,CK Hall,YQ Zhou.Protein Folding Pathways and Kinetics:Molecular Dynamics Simulations of B-strand Motifs[J].Biophysical Journal,2002(83):819-835.
[22] NV Dokholyan,SV Buldyrev,HE Stanley,et al.Discrete Molecular Dynamics Studies of the Folding of a Protein-like Model[J].Folding and Design,1998(3):577-587.
[23] NV Dokholyan,SV Buldyrev,HE Stanley,et al.Identifying the Protein Folding Nucleus Using Molecular Dynamics[J].Journal of Molecular Biology,2000(296):1183-1188.
[24] 蒋巍,钟方丽,史晋宜,等.生物膜胞外聚合物的研究进展[J].吉林化工学院学报.2001,18(3):85-88
[25] A Singh,S Snyder,L Lee,et al.Effect of Oligonucleotide Length on Theassembly of DNA Materials:Molecular Dynamics Simulations of Layer-by-layer DNA Films[J].Langmuir,2010(26):17339-17347.
[26] HP Schwefel.Numerical Optimization of Computer Models[M].John Wiley & Sons,Inc.,1981.
[27] Y Zhao.A Study on Fe2+- α-Helical-Rich Keratin Complex Formation Using Isothermal Titration Calorimetry and Molecular Dynamics Simulation[J].Journal of Pharmaceutical Sciences,2014(4):1224-1232.
[28] MS Saddala.Molecular dynamic simulations docking inhibitors of falcipain - 2[J].Online Journal of Bioinformatics,2014(1):98-105.
[29] E Temur,IH Boyacl, U Tamer, et al. A Highly Sensitive Detection Platform Based on Surface-enhanced Raman Scattering for Escherichia Coli Enumeration[J].Analytical and Bioanalytical Chemistry,2010(4):1595-1604.
[30] W Li,W Meng,P Tian.Impact of Stable Protein-protein Interaction on Protein Conformational Space[J].Chemical Research in Chinese Universities,2015(1):59-67.
[31] S Yan,CL.Suiter,G Hou,et al.Probing Structure and Dynamics of Protein Assemblies by Magic Angle Spinning NMR Spectroscopy[J].Chemistry & Physics of Lipids,2013(9):2047-2058.
[32] NF Brás,N Cerqueira,SF Sousa.Protein Ligand Docking in Drug Discovery[J].Protein Modelling,2014(1):249-286.
[33] A Johari,AA Moosavi-Movahedi,M Amanlou.Computational Investigation of Inhibitory Mechanism of Flavonoids as Bovine Serum Albumin Anti-glycation Agents[J].DARU Journal of Pharmaceutical Sciences,2014(22):79-86.
[34] AM Watkins,PS Arora.Structure-based Inhibition of Protein-protein Interactions[J].European Journal of Medicinal Chemistry,2015(13):480-488.
[35] M Levitt,S Lifson.Refinement of Protein Conformations Using a Macromolecular Energy Minimization Procedure[J].Journal of molecular biology,1969(46):269-279.
[36] M Avriel.Nonlinear Programming:Analysis and Methods[M].Courier Dover Publications,2012.

**吉林化工学院2013级本科生
Basic Principle of Molecular Dynamics and Application
in The Filed of Biologic Molecules Simulation
LIU Guan-chen
(College of Materials Science and Engineering,Jilin Institute of Chemical Technology,Jilin City 132022,China)
Abstract:This article briefly describes the molecular dynamics of development history,basic theory,basic steps and basic research as a means to carry out simulation in the field of application of biological macromolecules.
Key words:molecular dynamics;biologic high molecules
文章编号:1007-2853(2015)11-0117-03
通信作者:*路大勇,E-mail:dylu@jlict.edu.cn
作者简介:刘军伟(1985-),男,河南许昌人,吉林化工学院助教,在读博士,主要从事无机陶瓷材料方面的研究.
基金项目:吉林化工学院科技研究项目(2013107);吉林化工学院科技研究项目(2014046)
收稿日期:2015-08-27
中图分类号:Q 61
文献标志码:A DOI:10.16039/j.cnki.cn22-1249.2015.11.028
