B16N16Lin(n=1,2,3,4,6,12)团簇的电子结构及稳定性
2016-01-19陈宏善刘淑红
陈宏善,刘淑红
(西北师范大学物理与电子工程学院,
甘肃省原子分子物理与功能材料重点实验室,甘肃兰州 730070)
B16N16Lin(n=1,2,3,4,6,12)团簇的电子结构及稳定性
陈宏善,刘淑红
(西北师范大学物理与电子工程学院,
甘肃省原子分子物理与功能材料重点实验室,甘肃兰州730070)
摘要:基于密度泛函理论B3LYP/6-31G(d,p)方法,研究了Li原子与B16N16团簇的结合方式和稳定性,并分析了电子结构.结果表明,2个Li原子担载到1个B原子形成2个B-N键时最稳定,Li原子的平均结合能约为1.0 eV,大于Li原子之间的结合能,Li原子不会在团簇表面发生聚集.在B16N16Lin(n=1,2,3,4,6,12)体系中,Li原子向团簇转移电子,随着Li原子数目的增加,Li原子的净电荷由0.72 e减少到约0.50 e,Li原子的2s轨道形成的占据轨道不断升高,体系的能隙由2.64 eV减小到1.59 eV.
关键词:B16N16;B16N16Lin;稳定性;电子结构
收稿日期:2015-06-20;修改稿收到日期:2015-10-09
基金项目:国家自然科学基金资助项目(11164024)
作者简介:陈宏善(1963—),男,甘肃金塔人,教授,博士研究生导师.主要研究方向为原子与分子物理和材料粘弹性.E-mail:chenhs@nwnu.edu.cn
中图分类号:O 641.12
文献标志码:标志码:A
文章编号:章编号:1001-988Ⅹ(2015)06-0039-05
Abstract:Using density functional theory B3LYP/6-31G(d,p),the binding manners,stabilities and electronic structures of Li atoms on B16N16cluster are investigated.The results show that the most stable structure is a pair of Li atoms supported on two B-N bonds by one B atom.The binding energy of the Li atoms is about 1.0 eV which is greater than the binding energy between two Li atoms,so the Li atoms can not cluster on the surface of the cluster.The electron is transferred from the Li atoms to the cluster in the B16N16Lin(n=1,2,3,4,6,12) systems.With the increasing of the number of Li atoms,the net charge on the Li atoms is decreased from 0.72 e to 0.50 e.The occupied orbitals forming by Li 2s are pushed up gradually.The energy gap of the systems is reduced from 2.64 to 1.59 eV.
ElectronicstructuresandstabilityofB16N16Lin
CHENHong-shan,LIUShu-hong
(CollegeofPhysicsandElectronicEngineering,NorthwestNormalUniversity,
KeyLaboratoryofAtomicandMolecularPhysics&FunctionalMaterialsofGansuProvince,Lanzhou730070,Gansu,China)
Keywords:B16N16;B16N16Lincluster;stability;electronicstructure
BN单元与C2属于等电子体系,在发现各种碳纳米结构后,人们便预言存在类似BN体系的结构.1994年Rubio等发现氮化硼纳米管[1-2],同年Banhart[3]用强电子束照射无定形BN观测到卷曲的洋葱状BN团簇.1995年Boulanger等[4]用激光高温分解氨和三氯化硼的混合物,观测到多种近球状的BN纳米粒子.Stephan等[5]用强电子束照射BN纳米管和体材料在实验上得到了直径在0.4~0.7nm的笼状BN,特别稳定的幻数结构有B12N12,B16N16以及B28N28等.早在实验观察到BN笼状团簇之前,有关BN笼的理论研究就已报道.Xia等[6]根据C60的结构预测了B30N30的结构,因为经典的富勒结构有孤立五元环,因此不能避免同质键的存在.与C-C键相比,B-B键缺电子而N-N键富电子导致不稳定,为了完全消除B-B和N-N键最大化B-N键,团簇结构就必须由偶数元环(每个环中的原子数为偶数)组成.由此提出,稳定的(BN)n团簇应该由四元环与六元环交替式结构形成.Sun等[7]用AM1方法研究了不同的BN团簇,预言BnNn(n=12,16,20,28,36)具有幻数稳定性.Serfert和Folwer[8]对这些稳定的BnNn团簇的电子结构进行了更进一步的计算研究,与碳团簇相比,这些团簇具有较大的HOMO-LUMO能隙,说明氮化硼团簇比碳团簇有更高的稳定性.一些理论组得出了过渡金属原子修饰的富勒烯有很好的稳定性[9-11].武海顺基于密度泛函理论B3LYP方法,系统研究了掺杂过渡金属原子Cu,Ag,Au,Ni,Pd,Pt对B16N16笼的影响[12],表明掺杂后最高占据轨道和最低空轨道之间的能隙变窄,显示出金属的半导体性质.他们假设修饰的金属原子都是均匀地分布在主体材料上.但是,有人计算发现修饰的过渡金属原子在主体结构表面会发生聚集,这样将会明显地降低材料的稳定性[13-15],而如果用碱金属或碱土金属修饰则不会出现聚集现象[16-17].本文主要研究了Li原子担载到B16N16团簇上的几何结构和稳定性,并分析了其电子结构.
1理论方法
本文计算采用密度泛函理论(DFT)的B3LYP,电子交换能用HF和Becke的三参数杂化函形式[18],局部项修正使用VWNⅢ函数[19],非局部项用LYP泛函[20].所用基组是加入极化基的二重劈裂基组6-31G(d,p).对Li的不同担载位进行完全优化,得到了所有可能的稳定结构,并对能量较低结构进行了自然键轨道(NBO)[21]分析,最后给出了最稳定结构的态密度图[22].以上所有计算均在Gaussian03程序下完成.
2结果与讨论
2.1B16N16团簇的几何与电子结构
如图1所示,B16N16具有Td对称性,由12个六元环和6个四元环构成,每个四元环与4个六元环相接,48个B-N键分为两类:一类为四元环和六元环共用的R46键;另一类为六元环和六元环共用的R66键.B原子和N原子各有2种非等价位,一种是形成3个R66键,另一种是形成2个R46键和1个R66键.

图1 B16N16团簇最稳定结构
图2给出了B16N16价电子的总态密度和分态密度.价带分为两部分,其中较低的能带处在-26.96~-22.2eV内,主要来自N2s的贡献.较高的能带处在-16.78~-7.38eV内,主要由B的2s2p和N的2p轨道组成.NBO分析给出B原子上的净电荷为1.16e/1.18e,N原子上为-1.14e/-1.20e.N原子的2p轨道被来自B原子的电子填充,N的2p能级被提高.由于B原子和N原子的2s2p轨道介于sp2杂化和sp3杂化之间,从分态密度看出B的2p比2s的贡献大.

图2 B16N16团簇的总态密度和分态密度
图3给出了几个最高占据轨道和最低空轨道,最高占据轨道由垂直于笼表面N2p轨道形成,而较低的空轨道主要由垂直于笼表面B2p轨道组成.B16N16的能隙值为6.37eV,这是典型的BN体系的能隙.Li原子担载到B16N16团簇上时,将向团簇转移电子,上述空轨道的特征对于理解Li原子在B16N16团簇上的结合位是重要的.

图3 B16N16团簇的分子轨道(括号内数字代表简并度)
2.2Li担载B16N16的几何结构
为了确定Li原子担载在B16N16上不同位置的结合强度,定义单个Li原子的平均结合能Eb为
其中,E(B16N16Lin)和E(B16N16)分别为B16N16Lin和B16N16体系的能量,E(Li)为单个Li原子的能量.
考虑了B16N16结构中不等价的B原子和N原子顶位、B-N桥位以及环的上空位作为Li原子初始的结合位置.完全优化结果表明,担载1个Li原子时,位于R46桥位上最稳定,其结合能为0.55eV.担载2个Li原子时,除了对所有非等价结合位进行优化外,还考虑了不同自旋多重度.结果表明,当2个Li原子相隔较远时能量较高,Li原子倾向于以成对的形式结合在同1个B原子形成的B-N键上.对不同自旋多重度(1,3)进行优化表明,自选多重度为1时结合能比自旋多重度为3时低约0.35eV.如图4(a~c)所示,最稳定的结合位分别是2个Li原子位于同1个B原子形成的2个R46桥位、1个R46和1个R66桥位以及2个R66桥位,结合能分别为1.12,1.10和0.92eV.担载3个Li原子时,将所有桥位的不等价组合作为Li原子的初始结合位,优化后最稳定结构如图4(d,e)所示,当Li原子位于由同1个B原子形成的2个R46桥位、1个R66桥位和3个R66桥位时最稳定,结合能分别为1.08和1.02eV.当放入4个Li原子时,考虑了2对Li原子分别位于不同四元环的情况,优化后得到3种最稳定结构,如图4(f~h)所示,图4(f)和(g)结构中2对Li原子分别位于相对和相邻的2个四元环中,并且每个四元环中的2个Li原子位于同1个B原子形成的2个R46桥位上,结合能分别为1.10和1.09eV;图4(h)结构中4个Li原子分别位于同1个四元环上的2个N原子顶位和2个R66桥位,结合能为1.04eV.担载6个Li原子时考虑了3对Li原子在不同位置的组合,优化后得到3种最稳定结构,如图4(i~k)所示,图4(i,j)结构中6个Li原子分别位于图1中上半部的3个四元环中,并且每个四元环中2个Li原子位于同1个B相连的R46桥位上,结合能为1.07eV和1.06eV;图4(k)结构中2对Li原子位于上半部2个四元环中,1对位于下半部的1个四元环中,结合能为1.03eV.最后,计算了担载12个Li原子的情况,最稳定的结构如图4(l)所示,12个Li原子分别位于6个四元环中同1个B原子相连的2个R46桥位上,其中3对位于图中上半部分3个四元环中,另外3对位于图中下半部3个四元环中,结合能为0.96eV.表1列出了各结构的结合能,可以看出,2个或2个以上Li原子担载到B16N16上时,单个Li原子的平均结合能在0.92~1.12eV之间,基于相同的计算方法,Li2,Li3,Li4中每个Li原子的结合能分别为0.44,0.46,0.61 eV.显然,Li原子与B16N16团簇的结合能都大于Li原子之间的作用能,因此Li原子在B16N16团簇表面将会独立分布,而不会像过渡金属原子那样在主体表面发生聚集现象.表1还给出了Li-B和Li-N平均距离分别约为0.216和0.195nm.

图4 Li原子修饰的B16N16的稳定结构
2.3Li担载B16N16的电子结构
表1也给出了Li原子上的NBO(自然键轨道)电荷,当担载2个、4个、6个和12个Li原子时,最稳定结构中Li原子所带电荷分别为0.72e,0.70e,0.59e和约0.50e.当担载3个Li原子时,由于Li原子相互靠近,故所带电荷数减少,大约为0.45e.

表1 B16N16Lin体系中Li-B与Li-N平均距离、Li原子的
图5给出了B16N16Lin(n=1,2,3,4,6,12)体系的总态密度和Li的分态密度,并用虚线标注了最高占据轨道的位置.占据轨道分为3个能带,较低的2个能带和纯的B16N16一样,主要来自N的2s,B的2s2p以及N的2p.能量最高的能带由Li的2s组成,当n=2,4,6和12时,所包含的能级数目分别为1,2,3和6;当担载3个Li原子时,为开壳层结构,为便于比较,态密度取为α和β电子态密度和的一半.B16N16Lin(n=1,2,3,4,6,12)中,Li原子向B16N16转移电子,使得B、N原子中电子之间的排斥增大,较低的2个能带位置随Li原子数值增加略有上移.同样由于Li原子增加时由Li向团簇转移的电子数减少,由Li的2s形成的HOMO能级升高,带隙值逐渐减少.
3结论
基于密度泛函理论B3LYP/6-31(d,p),对Li担载的B16N16团簇结构进行优化,得到了B16N16Lin(n=1,2,3,4,6,12)的稳定结构.结果表明,Li原子在B16N16团簇表面的最佳结合方式是1对Li原子担载到同1个B原子形成的2个B-N桥位上.每个Li原子的平均结合能约1.10 eV;当6个四元环上担载12个Li原子时结合能为0.96 eV.基于相同的计算方法,Li2,Li3,Li4中单个Li原子的结合能分别为0.44,0.46,0.61 eV.因此Li原子在B16N16团簇表面不会发生聚集现象.通过对B16N16Lin(n=1,2,3,4,6,12)体系的态密度(DOS)和自然键轨道(NBO)分析,表明Li原子结合到B16N16团簇上时,向团簇转移电子,随着n的增加,Li原子上的净电荷由0.72 e减小为0.45 e,相应的HOMO能级由-4.11 eV升高到-2.82 eV,LUMO能级由-1.48 eV升高到-1.23 eV,体系的能隙值由2.64 eV逐渐变小到1.59 eV.
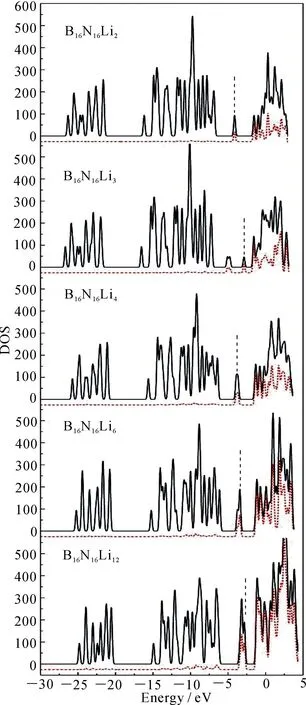
图中红色点划线表示Li的2s2p;虚线标注HOMO位置
参考文献:
[1]RUBIO A,CORKILL J L,COHEN M L.Theory of graphitic nitrde nanotubes[J].PhysRevB,1994,49:5081.
[2]BLASE X,RUBIO A,LOUIE S G,et al.Stability and band gap constancy of boron nitride nano tubes[J].EuroPhysLett,1994,28:335.
[3]BANHART F,ZWANGER M,MUHR H J.The formation of curled concentric-shell clusters in boron nitride under electron irradiation[J].ChemPhysLett,1994,231:98.
[4]BOULANGER L,ANDRIOT B,CAUCHETIER M,et al.Concentric shelled and plate-like graphitic boron nitride nano partieles produced by CO2laser pyrolysis[J].ChemPhysLett,1995,234:227.
[5]STEPHAN O,BANDO Y,LOISEAU A,et al.Formation of small single iayer and nested BN cages under electron irradiation of nanotubes and bulk material[J].ApplPhysA,1998,67:107.
[6]XIA X F,DANIEL A,JELSKI J R,et al.MNDO study of boron-nitrogen analogues of buekminster fullerene[J].JAmChemSoc,1992,114:6493.
[7]SUN M L,SLANINA Z,LEE S L,Square/hexagon route towards the boron-nitrogen clusters[J].ChemPhysLett,1995,233:279.
[8]SERFERT G,FOLWER P W,MITCHELL D,et al.Boron-nitrogen analogues of the fullerenes:electronic and structural properties[J].ChemPhysLett,1997,268:352.
[9]DURGUN E,JANG Y R,CIRACI S.Hydrogen storage capacity of Ti-doped boron-nitride and B/Be-substituted carbon nanotubes[J].PhysRevB,2007,76:3009.
[10]TAI T B,NGUYEN M T.A three-dimensional aromatic B6Li8complex as a high capacity hydrogen storage material[J].ChemCommun,2013,49:913.
[11]GIRI S,LUND F,NEZ A S,et al.Can star like C6Li6be treated as a potential H2storage material[J].JPhysChemC,2013,117:5544.
[12]CUI X Y,YANG B S,WU H S.Ab initio investigation of hydrogenation of(BN)16:A comparison with that of (BN)12[J].JMolStruct,(THEOCHEM) 2010,941:144.
[13]SHIN W H,YANG S H,GODDARD W A,et al.Ni-dispersed fullerenes:Hydrogen storage and desorption properties[J].ApplPhysLett,2006,88:053111.
[14]SUN Q,WANG Q,JENA P,et al.Clustering of Ti on a C60surface and its effect on hydrogen storage[J].JAmChemSoc,2005,127:14582.
[15]BARMAN S,SEN P,DAS G P.Ti-decorated doped silicon fullerene:Aprossible hydrogen-storage material[J].JPhysChemC,2008,112:19963.
[16]VENKATARAMANAN N S,BELOSLUDOV R V,NOTE R,et al.Theoretical investigation on the alkali-metal doped BN fullerene as a material for hydrogen storage[J].ChemPhys,2010,377:54.
[17]AN H,LIU C S,ZENG Z,et al.Li-doped B2C graphene as potential hydrogen storage medium[J].ApplPhysLett,2011,98:173101.
[18]BECKE A D.Density-functional thermochemistry.Ⅲ.The role of exact exchange[J].JChemPhys,1993,98:5648.
[19]VOSKOS H,WILK L,NUSAIR M.Accurate spin-dependent electron liquid correlation energies for local spin density calculations:A critical analysis[J].CanJPhys,1980,58:1200.
[20]LEE C,YANG W,PARR R G.Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J].PhysRevB,1988,37:785.
[21]WEIN H F.NBO3.0Program[M].3.0 ed.Madison,WI:University of Wisconsin Madison,2001.
[22]LU T,CHEN F W.Multiwfn:A multifunctional wave function analyzer[J].JCompChem,2012,33:580.
(责任编辑孙对兄)
