脱氢枞基α-氨基膦酸酯的合成*
2016-01-17沈明贵商士斌宋湛谦中国林业科学研究院林产化学工业研究所生物质化学利用国家工程实验室江苏南京20042中国林业科学研究院林业新技术研究所北京0009
沈明贵,王 丹,商士斌,宋湛谦(.中国林业科学研究院林产化学工业研究所生物质化学利用国家工程实验室,江苏南京 20042; 2.中国林业科学研究院林业新技术研究所,北京 0009)
脱氢枞基α-氨基膦酸酯的合成*
沈明贵1,2,王丹1,2,商士斌1,2,宋湛谦1
(1.中国林业科学研究院林产化学工业研究所生物质化学利用国家工程实验室,江苏南京210042; 2.中国林业科学研究院林业新技术研究所,北京100091)
摘要:以Yb(OTf)3为催化剂,聚氧乙基-α-生育酚癸二酸酯(PTS)为助溶剂,水为溶剂,经三组分[脱氢枞胺(1)、亚磷酸二乙酯(2)和芳醛(3a~3f)]缩合反应合成了6个含脱氢枞基的α-氨基膦酸酯衍生物(4a~4f),产率65%~75%,其结构经1H NMR,FT-IR和EI-MS确证。考察了溶剂和助溶剂对4a产率的影响。结果表明:在最优反应条件[以水为溶剂,1 1 mmol,2 1.2 mmol,3a 1 mmol,Yb(OTf)30.01 mmol,PTS 2 mL,于室温反应1 h]下,4a产率71%。Yb(OTf)3循环使用5次,4a产率64%。
关键词:脱氢枞胺;α-氨基膦酸酯;助溶剂;多组分合成
脱氢枞胺(1)是一种重要的松香衍生物[1],具有显著的抗溃疡[2]、抗菌[3]、抗焦虑[4]、抗病毒[5]、抗肿瘤[6]、细胞毒活性[7]和大电导钙激活钾离子通道开放调节性[8]等生物活性。因此,1被广泛应用于抗急性中风、癫痫、哮喘、高血压、胃蠕动过强和精神病等疾病药物的合成。
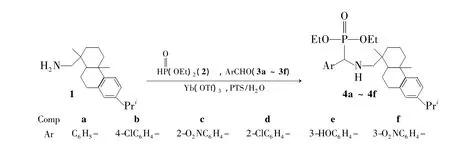
Scheme 1
α-氨基膦酸酯(Ⅰ)作为天然氨基酸类似物,
生物活性良好,在新药研发中具有重要意
义[9-11]。如Huang等[12]合成了含有脱氢枞基的
Ⅰ,抗肿瘤活性良好。根据结构拼合理论,将Ⅰ引入1的分子骨架中,可能获得具有更高生物活性的新化合物。
水作反应溶剂,具有廉价、无毒和易于操作的优点。Lipshutz's等通过在水中添加一种非离子型两亲分子——聚氧乙基-α-生育酚癸二酸酯(PTS),提高了有机分子在水相中的溶解度,于常温高效催化了一系列有机反应[13]。
本文在前期工作[14]的基础上,以Yb(OTf)3为催化剂,PTS为助溶剂,水为溶剂,经三组分[脱氢枞胺(1)、亚磷酸二乙酯(2)和芳香醛(3a~3f)]缩合反应合成了6个含脱氢枞基的α-氨基膦酸酯衍生物(4a~4f),产率65%~75%,其结构经1H NMR,FT-IR和EI-MS确证。考察了溶剂和助溶剂对4a产率的影响。
1 实验部分
1.1仪器与试剂
WRS-1B型熔点仪(温度未校正); Bruker Avance DMX-400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标); Bumem MB-154S型红外光谱仪(KBr压片); TSQ Quantum型液质联用仪。
1按文献[15]方法制备;其余所用试剂均为分析纯。
1.2 4a~4f的合成(以4a为例)
在反应瓶中加入2 ωt% PTS的水(2 mL)溶液,搅拌下依次加入3a 106 mg(1 mmol),1 286 mg(1 mmol),2 165 mg(1.2 mmol)和Yb(OTf)362 mg,于室温反应1 h(TLC检测)。反应物用乙酸乙酯(5 mL)萃取,水相(含催化剂)用乙酸乙酯(3×5 mL)洗涤后可直接用于下次反应;有机相用无水硫酸钠干燥,浓缩后经硅胶柱层析[洗脱剂:V(石油醚)∶V(乙酸乙酯)=9∶1]纯化得透明黏稠液体二乙基-苯基-脱氢枞胺基-甲基膦酸酯(4a)。用类似的方法合成透明液体4b~4f。
4a:产率71%;1H NMR δ:7.39(t,J=6.2 Hz,
2H),7.37~7.32(m,2H),7.28(t,J=6.2 Hz,1H),7.15(dd,J=8.1 Hz,3.2 Hz,1H),6.95(d,J=8.1 Hz,1H),6.85(s,1H); FT-IR ν:3 298,1 240,1 050 cm-1; EI-MS m/z:512.0[M+]。
4b:产率75%;1H NMR δ:7.46~7.38(m,
4H),7.16(d,J=8.2 Hz,1H),6.95(d,J=8.2 Hz,1H),6.86(s,1H); FT-IR ν:3 290,1 234,1 048 cm-1; EI-MS m/z:546.5[M+]。
4c:产率73%;1H NMR δ:7.93(t,J=10.9 Hz,2H),7.77(dd,J=13.3 Hz,6.8 Hz,1H),7.57(d,J=7.3 Hz,1H),7.16(dd,J=8.1 Hz,3.5 Hz,1H),6.95(d,J=8.1 Hz,1H),6.87(d,J=16.9 Hz,1H); FT-IR ν:3 300,1 250,1 055 cm-1; EI-MS m/z:557.0[M+]。
4d:产率70%;1H NMR δ:7.69(dd,J=15.5 Hz,7.7 Hz,1H),7.44(d,J=7.7 Hz,1H),7.38(t,J=7.6 Hz,1H),7.31(d,J=6.3 Hz,1H),7.19~7.10(m,1H),6.95(d,J=8.0 Hz,1H),6.86(d,J=10.7 Hz,1H); FT-IR ν:3 290,1 235,1 049 cm-1; EI-MS m/z:546.5[M+]。
4e:产率65%;1H NMR δ:7.68(s,1H),
7.15(dd,J=12.3 Hz,4.6 Hz,2H),7.22~7.06(m,2H),6.95(d,J=8.0 Hz,1H),6.86(s,1H),6.81(d,J=8.8 Hz,2H),6.69(t,J=6.8 Hz,1H),5.95(s,1H); FT-IR ν:3 480,3 295,1 238,1 050 cm-1; EI-MS m/z:528.0 [M+]。
4f:产率75%;1H NMR δ:8.33(s,1H),8.16(d,J=8.1 Hz,1H),7.90(d,J=7.1 Hz,1H),7.67(t,J=7.9 Hz,1H),7.15(t,J=6.8 Hz,1H),6.96(d,J=8.0 Hz,1H),6.85(d,J=10.8 Hz,1H); FT-IR ν:3 302,1 250,1 056 cm-1; EI-MS m/z:557.0[M+]。
2 结果与讨论
2.1合成方法
目前,合成4有两种方法。方法一:先合成亚胺;亚胺与亚磷酸酯经加成反应制得4[16];方法二:以醛、胺和磷酸酯为原料,经三组分“一锅煮”反应合成4[17]。根据绿色化学的发展需求,本文对方法2进行了改进,以Yb(OTf)3为催化剂,经三组分缩合反应合成了4。实验结果表明:该方法绿色环保,原子经济性高,副产物少,产率高,且催化体系可循环使用。
2.2条件优化
为优化反应条件,考察了溶剂和助溶剂对4a产率的影响,结果见表1。

表1 溶剂和助溶剂对4a产率的影响*Table 1 Effects of solvents and cosolvents on yield of 4a
由表1可见,单独以水作溶剂时,产率最低(21%,No.6);加入PTS后,产率提高至71%(No.7);继续增加PTS用量,产率基本不变(No.8)。以Triton X-100,SDS和TBAB为助溶剂时,产率分别为61%,62%和53%(No.9~No.11),但效果均不及PTS。因此,No.7为最优反应条件,即以水为溶剂,1 1 mmol,2 1.2 mmol,3a 1 mmol,Yb(OTf)30.01 mmol,PTS 2 mL,于室温反应1 h。
2.3循环性能
PTS水溶性较好,在部分有机溶剂(如:烷烃、乙醚、乙酸乙酯等)中溶解性较差。因此,利用这一特性,Yb(OTf)3可只经简单相分离和洗涤即可直接用于下一循环4a~4f的合成。以4a的合成为例,考察了Yb(OTf)3的循环性能,结果见表2。

表2 Yb(OTf)3的循环性能*Table 2 Cycle performance of Yb(OTf)3
由表2可见,Yb(OTf)3经5次循环使用,4a产率基本没有降低;当第6次循环使用后,产率略有降低,这可能是由于Yb(OTf)3反复洗涤回收造成的损失所致。
3 结论
合成了6个含脱氢枞基的α-氨基膦酸酯衍生物(4a~4f)。在最优反应条件[以水为溶剂,1 1 mmol,2 1.2 mmol,3a 1 mmol,Yb(OTf)30.01 mmol,PTS 2 mL,于室温反应1 h]下,4a产率71%。Yb(OTf)3循环使用5次,4a产率64%。
参考文献
[1]周志,林中祥.N,N-四氯邻苯二甲酰基-13-硝基-7,7-C60-脱异丙基脱氢枞胺的合成[J].合成化学,2014,22(1):63-67.
[2]Wada H,Kodato S,Kawamori M,et al.Antiulcer activity of dehydroabietic acid derivatives[J].Chem Pharm Bull,1985,33:1472-1487.
[3]Gigante B,Silva A M,Marcelo C M J,et al.Structural effects on the bioactivity of dehydroabietic acid derivatives[J].Planta Med,2002,68:680-684.
[4]Tolmacheva I A,Tarantin A V,Boteva A A,et al.Synthesis and biological activity of nitrogen-containing derivatives of methyl dehydroabietate[J].Pharm Chem J,2006,40:489-493.
[5]Tagat J R,Nazareno D V,Puar M S,et al.Synthsis and anti-herpes activity of some A-ring functionalized dehydrroabietane derivatives[J].Bioorg Med Chem Lett,1994,4:1101-1104.
[6]Kinouchi Y,Ohtsu H,Tokuda H,et al.Potential antitumor-promoting diterpenoids from the stem bark of piceaglehni[J].J Nat Prod,2000,63:817-820.
[7]Prinz S,Mullner U,Heilmann J,et al.Oxidation products of abietic acid and its methyl ester[J].J Nat Prod,2002,65:1530-1534.
[8]Ohwada T,Nonomura T,Maki K,et al.Dehydroabietic acid derivatives as a novel scaffold for large-conductance calcium-activated K+channel openers[J].Bioorg Med Chem Lett,2003,13:3971-3974.
[9]Lindsay B S,Barrows L R,Cop B R.Structural requirements for biological-activity of the marine alkaloid ascididemin[J].Bioorg Med Chem Lett,1995,5:739-742.
[10]Maier L,Diel P J.Organic phosphorus compounds 941 preparation,physical and biological properties of amino-arylmethylphosphonic and phosphonous acids[J].Phosphorus,Sulfur Silicon Relat Elem,1991,57:57-64.
[11]Alien J G,Atherton F R,Hall M J,et al.Phosphonopeptides:A new class of synthetic antibacterial agents[J].Nature,1978,272:56-58.
[12]Huang X C,Wang M,Pan Y M,et al.Synthesis and antitumor activities of novel thiourea α-aminophosphonates from dehydroabietic acid[J].Eur J Med Chem,2013,69:508-520.
[13]Lipshutz B H,Ghorai S,Leong W W Y,et al.Manipulating micellar environments for enhancing transition metal-catalyzed cross-couplings in water at room temperature[J].J Org Chem,2011,76:5061-5073.
[14]Shen M G,Shang S B,Song Z Q,et al.Highly efficient one-pot synthesis of α-aminophosphonates catalyzed by ytterbium triflate in water[J].Syn Commun,2014,44:361-367.
[15]Gottstein W J,Cheney L C.Dehydroabietylamine:A new resolving agent[J].J Org Chem,1965,30(6):2072-2073.
[16]李在国,黄润秋,杨昭,等.含苯并噻唑杂环的α-氨基膦酸二苯酯的合成及生物活性[J].应用化学,1999,16:90-92.
[17]Ambica K S,Taneja S C,Hundal M S,et al.Onepot synthesis of α-aminophosphonates catalyzed by antimony trichloride adsorbed on alumina[J].Tetrahedron Lett,2008,49:2208-2212.
·制药技术·
·快递论文·
Synthesis of α-Aminophosphonate
Derivatives Containing Dehydroabietic Group
SHEN Ming-gui1,2,WANG Dan1,2,SHANG Shi-bin1,2,SONG Zhan-qian1
(1.National Engineering Laboratory for Biomass Chemical Utilization,Institute of
New Technology of Forestry,Institute of Chemical Industry of Forest Products,Nanjing 210042,China;
2.Institute of New Technology of Forestry,Institute of Chemical Industry of Forest Products,Beijing 100091,China)
Abstract:Six α-aminophosphonates containing dehydroabietic group(4a~4f),in yield of 65%~75%,were synthesized by a three-component condensation reaction from dehydroabietylamine(1),diethyl phosphite(2)and aromatic aldehyde(3a~3f),using water as the solvent,ytterbium triflate [Yb(OTf)3]as the catalyst and polyoxyethanyl α-tocopheryl sebacate(PTS)as the cosolvent.The structures were comfirmed by1H NMR,FT-IR and EI-MS.The effects of solvents and cosolvents on the yield of 4a were investigated.The results showed that the optimum reaction conditions of synthesis 4a at room temperature for 1 h were as follows:1 1 mmol,2 1.2 mmol,3a 1 mmol,Yb(OTf)30.01 mmol,PTS 2 mL,using water as the solvent.The yield of 4a was 71% under the optimum conditions.The yield of 4a was 64% after Yb(OTf)3cycled for five times.
Keywords:dehydroabietylamine;α-aminophosphonate; cosolvent; multiple component reaction synthesis
通讯作者:商士斌,博士生导师,Tel.025-85482499; E-mail:shangsb@ hotmail.com
作者简介:沈明贵(1982-),男,汉族,江苏溧阳人,博士,主要从事生物质资源化学与应用的研究。
基金项目:国家自然科学基金云南联合基金资助项目(U1202265)
收稿日期:2014-08-20;
修订日期:2015-04-23
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.06.0503 *
文献标识码:A
中图分类号:O621.3
