Development of Neural Network for BLSOM Clustering of HA Genes of Avian Influenza Viruses Isolated in Guangdong Province
2016-01-12,,,,,,,
, , , , , , ,
Guangdong Inspection and Quarantine Technology Center/Guangdong Provincial Key Laboratory of Animal and Plant and Food Import and Export Technology/AQSIQ State Key Laboratory of Avian Influenza, Guangzhou 510623, China
1 Introduction
Avian influenza can cause great economic losses to poultry industry, and according to statistics of OIE, H5N1avian influenza infected 668 humans, and the mortality rate reached 58.83%. Currently, the emerging H7N9avian influenza continues to be prevalent, and there are new human cases, making it more difficult to prevent and control avian influenza. In fact, the zoonotic influenza virus is spread to humans ultimately, and brings a great threat to public health[1]. In the avian influenza virus monitoring, with the rapid progress of high-throughput sequencing technology, the gene pool data increase sharply, making the classic phylogenetic tree analysis difficult, so there is a need to conduct technological innovation. Pearl River Delta is located in the international migratory routes of migratory birds, and has a mild and humid climate. It is the world’s avian influenza outbreak center. Survey shows that the avian influenza infection is increasingly serious in the live poultry markets of Guangzhou, Jiangmen, Zhaoqing and other places, and these markets become important repository of virus[2-4]. The positive rate reaches 32.73%, and it is even up to 75% on the chopping board for the slaughter[5]. It is also found that it is dominated by H9subtype, and there are also H5and H7subtypes, greatly different from the vaccine strains[6]. At the same time, the majority of residents are often in contact with live poultry, and humans will be in critical condition or even die after infection[7-8]. Therefore, it is of great significance to study the classification methods for avian influenza virus.
2 Materials and methods
2.1DatasourcesAll strains used in this study came from Guangdong Province, providing all of the HA gene sequences. Among them, H1N1influenza virus was isolated from the pig strains by South China Agricultural University and from the human strains by the medical unit; H3N2strains were viral samples isolated by Guangdong Provincial Center for Disease Control from the patient; H5N1viruses were the poultry strains isolated by Harbin Veterinary Research Institute and South China Agricultural University, and human infections isolated by National Influenza Center; H7N9viruses were the recent epidemic strains, provided by Guangdong Provincial Center for Disease Control, 3 isolated from humans and 2 from chickens; H9N2was isolated from chickens by Harbin Veterinary Research Institute and South China Agricultural University.
2.2Researchmethods
2.2.1BLSOM algorithm. We established BLSOM (Batch-Learning Self-Organizing Map) artificial neural network to receive the external input to produce different response regions and simulate human brain’s self-organizing learning process[9]. The Euclidean distance was calculated as follows:
2.2.2Data normalization. We measured the number of HA gene fragments for each strain[10], and the normalization formula was as follows:
2.2.3MATLAB realization. The programming was based on Reference[11], and MATLAB (2014) was run. Part of the program codes are as follows:
fx>>
%% clear environment variable
clc
clear
%% input data
% load data
load (’c:data.mat’);
P=data;
……
The functionnewsomwas used to establish SOM network, and the competition layer consisted of 6×6=36 neurons. The functionstrainandsimwere used for training and simulation; the functionplotsomwas used to draw the variable relation diagram; the functionvec2indwas used to convert data.
Table1BLSOMcharacteristicgeneticfragmentstatisticsabouttheavianinfluenzavirusprevalentinGuangdongProvince
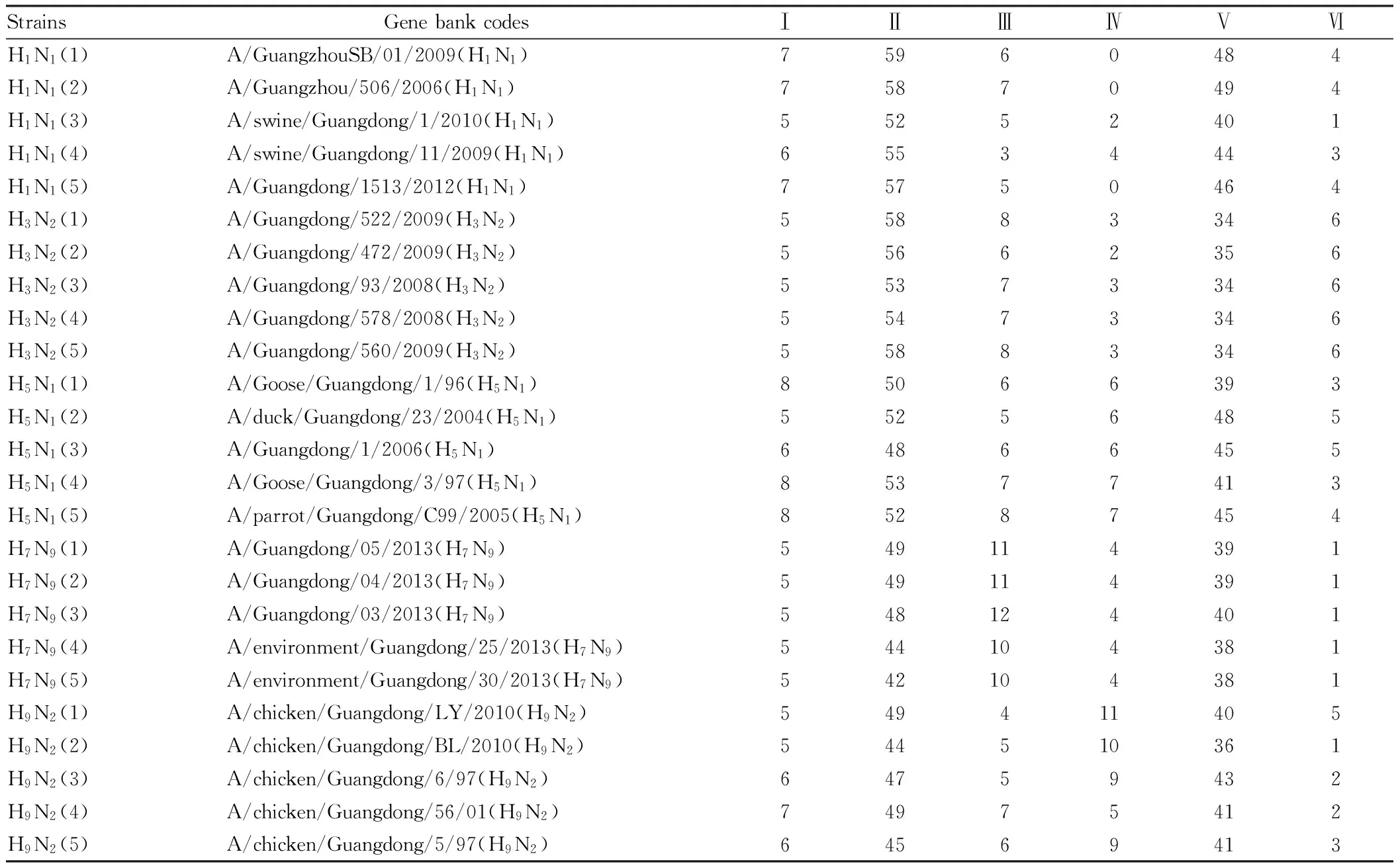
StrainsGenebankcodesⅠⅡⅢⅣⅤⅥH1N1(1)A/GuangzhouSB/01/2009(H1N1)75960484H1N1(2)A/Guangzhou/506/2006(H1N1)75870494H1N1(3)A/swine/Guangdong/1/2010(H1N1)55252401H1N1(4)A/swine/Guangdong/11/2009(H1N1)65534443H1N1(5)A/Guangdong/1513/2012(H1N1)75750464H3N2(1)A/Guangdong/522/2009(H3N2)55883346H3N2(2)A/Guangdong/472/2009(H3N2)55662356H3N2(3)A/Guangdong/93/2008(H3N2)55373346H3N2(4)A/Guangdong/578/2008(H3N2)55473346H3N2(5)A/Guangdong/560/2009(H3N2)55883346H5N1(1)A/Goose/Guangdong/1/96(H5N1)85066393H5N1(2)A/duck/Guangdong/23/2004(H5N1)55256485H5N1(3)A/Guangdong/1/2006(H5N1)64866455H5N1(4)A/Goose/Guangdong/3/97(H5N1)85377413H5N1(5)A/parrot/Guangdong/C99/2005(H5N1)85287454H7N9(1)A/Guangdong/05/2013(H7N9)549114391H7N9(2)A/Guangdong/04/2013(H7N9)549114391H7N9(3)A/Guangdong/03/2013(H7N9)548124401H7N9(4)A/environment/Guangdong/25/2013(H7N9)544104381H7N9(5)A/environment/Guangdong/30/2013(H7N9)542104381H9N2(1)A/chicken/Guangdong/LY/2010(H9N2)549411405H9N2(2)A/chicken/Guangdong/BL/2010(H9N2)544510361H9N2(3)A/chicken/Guangdong/6/97(H9N2)64759432H9N2(4)A/chicken/Guangdong/56/01(H9N2)74975412H9N2(5)A/chicken/Guangdong/5/97(H9N2)64569413
Note: The characteristic genetic fragments were I.GGGG, II.AAA, III.TTTC, IV.TCTT, V.AAG, VI.ACGG, respectively.
3 Results and analysis
3.1MicroorganismBLSOMclassificationAt present, microbial genomic information has increased significantly, and there is a need for new technical means to conduct a comprehensive analysis. The common GC value analysis method for microbial genome is simple, not suitable for processing large amounts of genomic information, and the results can not reflect the essential characteristics of microbial genetic variation. The non-sequence-alignment self-organizing map (SOM) and the improved BLSOM method, can be used to classify the 1 kb genetic fragment and predict the direction of variation. BLSOM uses the visual classification tool of advanced computer software which can reveal virus host-dependence and codon bias caused by natural selection, to identify high-risk types in millions of microbial gene data and monitor high-risk strains. It is of great significance to biomedicine and preventive veterinary medicine. The new MATLAB software toolbox provides neural network function, and can simulate the human brain to complete BLSOM competitive learning and training, pattern recognition, classification and identification. It is widely applied in engineering, finance, agriculture, environmental protection, education, public security and a variety of scientific studies[11-12]. In this paper, it was used to study the avian influenza virus and achieved initial success, worthy of further study.
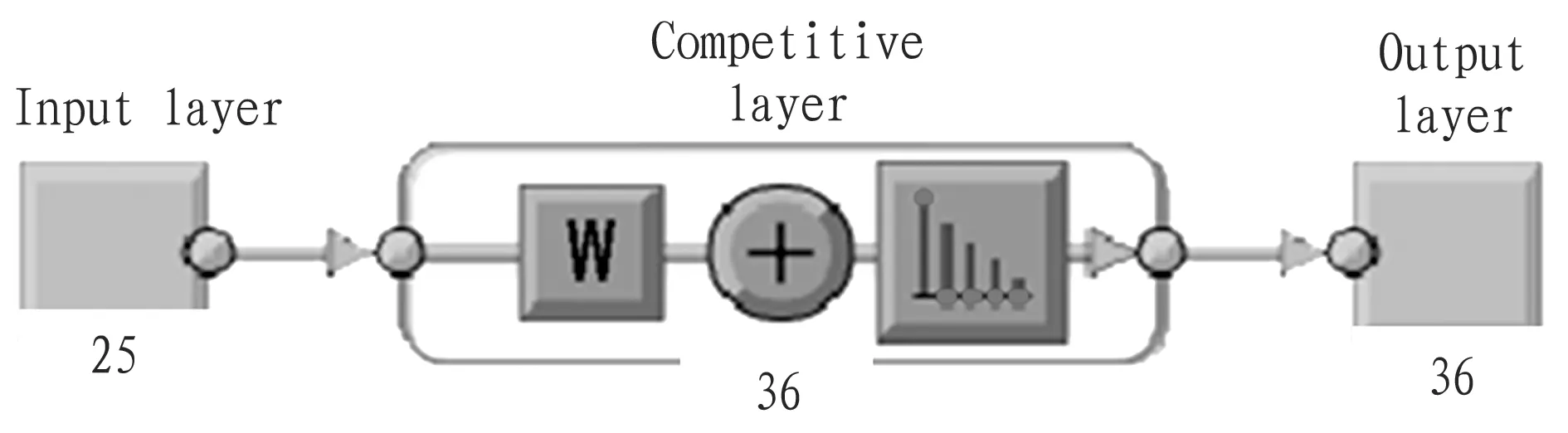
Fig.1BLSOMalgorithmflowchartforHAgenesofavianinfluenzavirusisolatedinGuangdongProvince
3.2AvianinfluenzaclassificationstandardandBLSOMclassificationAccording to statistics, the current nucleic acid sequences about avian influenza virus in the gene pool have been as many as 730000, and the number for H1N1, H3N2, H5N1and H9N2is 110000, 83000, 27000 and 14000, respectively. The conventional evolutionary tree and other analysis tools fail to see the whole picture[13]. BLSOM method can handle more than one million gene sequences at the same time, and the analysis results are consistent with the evolutionary tree. At the genetic level and in the oligonucleotide (2-4 bp) fragment composition, avian influenza shows significant host dependence, namely the self-organization and classification characteristics based on host, which is the biological basis of BLSOM classification. The influenza virus growth depends on many host factors such as nucleotides, amino acids and tRNA, while avoiding the antiviral mechanism of host such as the action of antibodies, cytotoxic T cells, interferon and RNA interference, thereby forming unique host dependence of gene structure. However, we often can not draw the conclusion of host dependence from the single nucleotide BLSOM, and the tetranucleotide BLSOM classification effect based on host is good. The results from Table 2, Fig. 3, Table 3 show that except HA genes, all 8 genetic fragments can be used for BLSOM analysis.
Fig.2DistancebetweenadjacentBLSOMneuronsforsomeavianinfluenzavirusstrainsprevalentinGuangdong
Table2BLSOMtrainingstepsandclusteringresultsforsomeavianinfluenzavirusstrainsprevalentinGuangdong

NumberoftrainingstepsClusteringresults103614363613031112362565036123352411001335651936200211213143650033962425110001122831936

Fig.3BLSOMclusteringmapforsomeavianinfluenzavirusstrainsprevalentinGuangdong
Table3BLSOMclusteringresultsforsomeavianinfluenzavirusstrainsprevalentinGuangdong

TypeNameofthestrain1H1N1(1),H1N1(2),H1N1(4),H1N1(5)2H3N2(1),H3N2(2),H3N2(3),H3N2(4),H3N2(5)3H5N1(1),H5N1(2),H5N1(3),H5N1(4),H5N1(5)4H7N9(1),H7N9(2),H7N9(3),H7N9(4),H7N9(5)5H9N2(1),H9N2(3),H9N2(4),H9N2(5)6H1N1(3),H9N2(2)
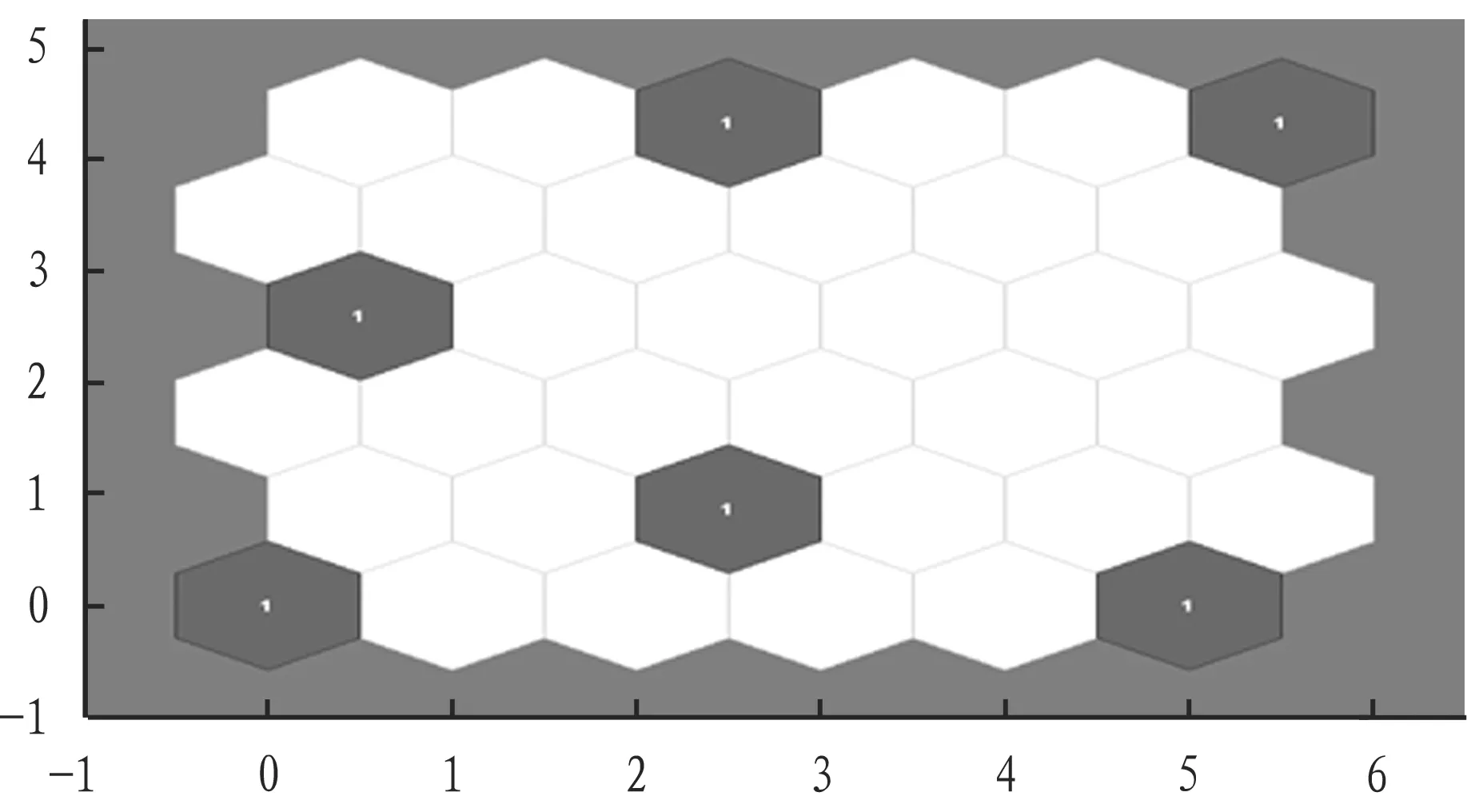
Fig.4BLSOMmapforthetestsamplesconcerningHAgenesofavianinfluenzavirusisolatedinGuangdongProvince
3.3BLSOMsoftwaretool,parameterandshortnucleotidefragmentselectionThe natural selection of avian influenza virus gene codon has pressure on all 8 fragments which can be used for BLSOM analysis to obtain genetic characteristics and host preference information. Tetra-BLSOM can clearly divide the human influenza virus from swine influenza virus according to region and color, and the high-risk strain located in the border between two regions due to the variation can be identified. From Fig. 3, we can clearly see that the strain structure of H3N2and H7N9is consistent, and there is strain variation for H9N2, H5N1and H1N1. In this study, 6 kinds of oligonucleotide fragments are selected for BLSOM analysis, and the main avian influenza strains can be identified. When the number of training steps is more than 100, the effective clustering effect is generated (Table 2). The neurons in the upper right corner are distant (Fig. 2), and the test samples are evenly distributed (Fig. 4). The methods and softwares that can handle large data sequences simultaneously are for further study. BLSOM is used to analyze the host dependence on oligonucleotide, identify sequence variation direction in huge gene data, and monitor high-risk strains of animals and humans, thus it is an important issue on viral molecular evolution study[9]. In this study, there is large variation in H5N1, and BLSOM map is significantly different (Fig. 3). There is a need to conduct further studies on the use of BLSOM as virus warning and traceability tool.
[1] SONG JD, ZHU DG, YUAN LP,etal. Etiology analysis of avian influenza pathogen from the live poultry market in Jiangmen Region during 2011-2013[J]. Guangdong Journal of Animal and Veterinary Science,2014,31(6):6-9. (in Chinese).
[2] LU QF, CAO JW, FENG XH,etal. Etiology analysis of avian influenza pathogen from the live poultry market in Jiangmen Region during 2011-2013[J]. Guangdong Journal of Animal and Veterinary Science,2014,39(4):18-19. (in Chinese).
[3] LU EJ, CHEN YY, LIU JW,etal. Surveillance analysis on the occupational population exposed to avian influenza virus and contamination in market environment of Guangzhou city in 2013[J]. Chinese Journal of Pest Control,2014,30(9):980-984.(in Chinese).
[4] LU JY, LU EJ, LI KB,etal. Analysis on avian influenza surveillance among occupational population in poultry environment in Guangzhou from 2011 to 2012[J]. Chinese Journal of Pest Control,2013,29(6):591-593.(in Chinese).
[5] ZHU BL, HUANG GH, MAI W,etal. Surveillance analysis on the high risk population and environment of avian influenza in Zhaoqing, 2011-2012[J]. Journal Of Tropical Medicine,2014,14(1):115-117. (in Chinese).
[6] LI GW, YAN ZQ, LIAO CT,etal. Phylogenetic analysis of hemagglutinin genes of H9N2subtype avian influenza viruses isolated in Guangdong and Guangxi provinces during 2011-2012[J]. Chinese Journal of Veterinary Science,2014,34(3):461-464. (in Chinese).
[7] CHEN B, MA ZC, RAO DP,etal. Epidemiological investigation on the cases of human infection With H7N9avian influenza in Shenzhen[J]. The Journal of Medical Theory and Practice,2014,27(21):2924-2925.(in Chinese).
[8] KONG DF, QIN YM, MEI SJ,etal. Epidemiological analysis on 2 cases infected with highly pathogenic human avain influenza in Shenzhen[J]. Chinese Journal of Pest Control,2013,29(12):1390-1392.(in Chinese).
[9] IWASAKI Y, ABE T, WADA K,etal. Prediction of directional changes of Influenza A virus genome sequences with emphasis on pandemic H1N1/09 as a model case[J]. DNA Research, 2011, 18(2): 125-136.
[10] IWASAKI Y, ABE T, WADA Y,etal. Novel bioinformatics strategies for prediction of directional sequence changes in influenza virus genomes and for surveillance of potentially hazardous strains[J]. BMC Infectious Diseases,2013,13:386.
[11] WANG XC, SHI F, YU L,etal. Analysis on 43 cases about MATLAB neural network[M]. Beijing:Beihang University Press,2013. (in Chinese).
[12] ZHANG XR, ZHANG YL, LIU LS,etal. Zoning by land types based on SOFM network:A case study on transect of eastern Tibetan Plateau[J]. Geographical Research,2013,32(5):839-847.(in Chinese).
[13] SONG QQ, CHAI ZX, ZHONG JC,etal. Codon usage bias and cluster analysis on genes of avian influenza virus[J]. Biotechnology,2014,24( 2):48-53.(in Chinese).
杂志排行

Asian Agricultural Research的其它文章
- Innovation on Distant Hybridization of Saline-tolerant Mud Flat Spartina and Rice Germplasm
- Advances in Studies of Genetic Improvement of Sugarcane
- Necessities and Practical Approaches for Beautiful Countryside Construction
- A Study of the Issues concerning Development of China’s Agricultural Product Logistics
- Review of Agricultural Insurance Development in the New Period in China
- A Study of the Factors that Affect Safe Full Heading of Machine-transplanted Rice Seedlings in the Temperate and Cool Rice-growing Areas