高脂饲粮对日本鹌鹑(Cotur nix japonica)盲肠微生物群落结构的影响
2016-01-08刘莎莎张宏福郑明德计
刘莎莎张宏福郑明德计 成
(1.中国农业科学院北京畜牧兽医研究所,动物营养国家重点实验室,北京100193;2.中国农业大学动物科学技术学院,北京100193;3.加拿大英属哥伦比亚大学,禽类研究中心,温哥华V6T1Z4)
高脂饲粮对日本鹌鹑(Cotur nix japonica)盲肠微生物群落结构的影响
刘莎莎1,2张宏福1∗郑明德3计 成2
(1.中国农业科学院北京畜牧兽医研究所,动物营养国家重点实验室,北京100193;2.中国农业大学动物科学技术学院,北京100193;3.加拿大英属哥伦比亚大学,禽类研究中心,温哥华V6T1Z4)
摘 要:本试验旨在研究高脂饲粮对日本鹌鹑(Cotur nix japonica)盲肠微生物群落结构、组成及其脂肪代谢的影响。选择160只1日龄日本鹌鹑,1~6周龄饲喂半合成饲粮,6周龄时,所有鹌鹑随机分成2个组,每组8个重复,每个重复10只。2组分别饲喂半合成饲粮和添加胆固醇的半合成饲粮,继续饲喂6周。试验期结束后,每个重复随机挑选1只接近平均体重的鹌鹑进行屠宰,收集血液分离得到血浆用于脂肪代谢指标分析;采集鹌鹑盲肠内容物,用焦磷酸测序分析微生物群落结构和组成。结果表明:1)高脂饲粮组鹌鹑总胆固醇(TC)、低密度脂蛋白(LDL)含量和LDL/高密度脂蛋白(HDL)显著提高(P<0.05);2)高脂饲粮显著降低盲肠微生物多样性(P<0.05);在微生物科水平,高脂饲粮显著降低Lactobacillal、Streptococcaceae和Clostridiales (P<0.05)、其他(others)科的丰度(P<0.05),显著提高Erysipelotrichales、Erysipelotrichaceae科的丰度(P<0.05),在微生物属水平,高脂饲粮显著降低Ruminococcaceae、Ruminococcus的丰度(P<0.05),但是显著提高了Erysipelotrichaceae、cc_115和Erysipelotrichaceae、Eubacterium的丰度(P<0.05);3)cc_115(Erysipelotrichaceae)和未被分类的微生物(属于Lachnospiraceae科)丰度与TC、LDL含量和LDL/HDL呈显著正相关(P<0.05),除此之外Eubacterium(Erysipe⁃lotrichaceae)只与LDL/HDL呈显著正相关(P<0.05),Ruminococcus(Ruminococcaceae)的丰度与TC、LDL含量和LDL/HDL呈显著负相关(P<0.05),Ruminococcus(Lachnospiraceae)的丰度与HDL含量呈显著正相关(P<0.05)。由此可见,高脂饲粮影响了日本鹌鹑盲肠微生物群落的结构、组成及其脂肪代谢。
关键词:高脂饲粮;肠道微生物群落;脂肪代谢
自从2005年新一代测序技术(next⁃generation sequencing,NGS)问世以来,对科学研究领域产生了翻天覆地的影响。随着核酸序列数据的单碱基测序费用下降,给动物科学研究带来了更多的新思路和新方案,目前已经广泛应用于动物全基因组测序、基因组重测序、转录组测序、小RNAs测序和微生物测序等方面。微生物在动物肠道能降解食物中纤维,合成维生素帮助动物吸收营养物质,维持肠道免疫系统功能,抵挡有害微生物的侵入。Zhao等[1]用新一代高通量测序技术研究鸡双向选择家系的个体肠道微生物,提出肠道微生物菌群的组成和分布具备数量性状的特点,将其视为宿主的一个性状。动物肠道中寄生的微生物群落种类繁多,数量惊人,是动物体细胞总量的10倍以上[2],目前依然有80%以上的微生物不为所知,宿主和肠道微生物相互作用的特点和规律还不是很清楚。有研究发现,饲喂高脂肪饲粮给无菌小鼠和正常小鼠,无菌小鼠没有得肥胖症;进一步研究分别移植肥胖症小鼠和瘦鼠的肠道微生物到无菌小鼠肠道中发现,移植肥胖小鼠肠道微生物的无菌小鼠会得肥胖症,而对照组无菌小鼠正常[3-4]。这些结果说明肠道微生物能调节宿主代谢和炎症反应,肠道微生物与宿主营养和免疫关系的研究在世界范围内越来越受到瞩目。高脂饲粮会引起宿主机体的异常代谢,产生炎症反应,与肠道微生物群落功能改变密切相关,因为微生物在动物肠道中保持一种平衡,当肠道微生物在宿主体内的平衡被打破,发生紊乱时,会对动物的生长和健康产生影响[5],了解微生物群落功能的前提是掌握其结构和组成。随着科学技术的不断更新,NGS的高通量数据、高检测深度和高准确率等优点可以解决以往研究微生物试验方法的不足,如变性梯度凝胶电泳(DGGE)分辨率低,且不能反映微生物的丰度情况;生物芯片只能检测到已知微生物,没有办法探测出未知微生物的情况。使用NGS使研究者能够准确检测到肠道微生物的丰度和组成、不同个体肠道微生物的共同及其独有特征,肠道微生物组成对肠道群落功能的预测等[6]。目前关于肠道微生物与宿主代谢关系研究主要使用小鼠模型,本研究使用日本鹌鹑(Cotur⁃nix japonica)作为动物模型,通过饲喂高脂饲粮和对照饲粮,研究其对宿主盲肠微生物群落结构、组成及其脂肪代谢的影响,为探索肠道群落在宿主能量和脂肪代谢方面所起的作用提供依据。
1 材料与方法
1.1 试验设计及样品采集
选择1日龄日本鹌鹑160只,1~6周龄饲喂半合成饲粮,6周龄时,所有鹌鹑随机分成2个组,每个组8个重复,分别饲喂半合成饲粮(对照饲粮)和添加胆固醇的半合成饲粮(高脂饲粮),继续饲喂6周。半合成试验饲粮配制根据NRC(1994)鹌鹑营养需要标准。试验期间动物在同一标准下养殖,自由采食和饮水。试验期结束时,每个重复随机挑选1只接近平均体重的鹌鹑进行屠宰,收集血液分离得到血浆,用镊子和剪刀快速采集盲肠及内容物放到离心管中,干冰速冻,-20℃保存。所有试验用具都经过高压灭菌处理,试验台用75%酒精擦拭。

表1 试验饲粮组成及营养水平(风干基础)Table 1 Composition and nutrient levels of experimental diets(air⁃dry basis) %
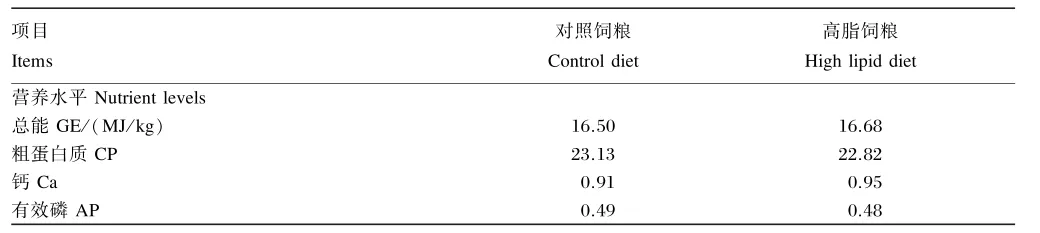
续表1
1.2 测定指标与方法
1.2.1 血浆脂肪代谢指标测定
血浆脂肪代谢指标有总胆固醇(TC)、高密度脂蛋白(HDL)、低密度脂蛋白(LDL)、甘油三酯(TGs)。TC、HDL和TGs含量采用酶法测定使用ADVIA 1650化学分析仪,HDL含量采用直接测定法[7-9]。LDL含量使用Friedewald公式[10-11],利用TC、HDL和TGs含量的测定值计算得出。1.2.2 盲肠食糜总DNA的提取及焦磷酸测序
将盲肠内容物轻轻挤到离心管,食糜总DNA的提取按照PowerMax Soil DNA Isolation(Mo.Bio laboratories.Inc.,Carlsbad,CA)试剂盒中的说明进行。PCR扩增采用FastStart High Fidelity体系(Roche Molecular Diagnostics,Branchburg,NJ,USA),341F(5’-ACTCCTACGG GAGGCAGCAG-3’)和926R(5’-CCGTCAATTCMTTTGAGTTT-3’)引物扩增微生物16s DNA V3~V5区。引物341F和926R分别添加适配序列(adaptor A,B),同时926R引物添加上不同的识别序列(a multi⁃plex identifier sequences,MID)。使用454 GS Jun⁃ior平台(454 Life Sciences⁃a Roche Company,Branford,CT,USA)对PCR产物进行测序。
1.3 数据分析
1.3.1 生物信息分析流程
QIIME(quantitative insights into microbial ecol⁃ogy)软件包对测序结果进行修剪降噪(拼接和去除引物)、筛选(质量控制)、匹配和分析[12]。满足如下条件的数据序列被剔除:数据平均质量得分≤25;序列长度<150 bp或者>900 bp;没有引物的序列、错误及包含模糊不清的序列;同聚体超过8 nt。数据去噪使用安装在QIIME的DENOISING v.0.9.1[13]软件。嵌合体序列剔除使用Chimera Slayer。数据根据各自的识别序列进行分组。UCLUST(http://www.drive5.com/usearch/)将含有超过97%的相同序列被分到同一个操作分类单元(operational taxonomic units,OTUs)。代表序列(representative sequences)是OTUs中最丰富的序列。使用Ribosomal Database Project(RDP)分类软件2.0.1[14]对代表序列(97%的相似)进行匹配分类。使用PyNAST对OTUs进行校准,要求最低校准长度是150 bp,序列75%以上相似[15]。然后使用PHLANE mask(http://greengenes.lbl.gov/)去除变异区段。
1.3.2 统计数据分析
使用SPSS 13.0对血浆中脂肪代谢指标及微生物的门、科和属的丰富度进行单因素方差分析(one⁃way ANOVA),数据用平均值±标准误表示,置信区间是95%。PC⁃ORD用于主成分分析(principal component analysis,PCA)分析盲肠微生物群落β多样性和PERMANOVA分析[16-17]。热图(heatmap)分析微生物的丰度与血浆中脂肪代谢指标的相关性。
2 结 果
2.1 鹌鹑血浆中脂肪代谢指标
由表1所示,鹌鹑血浆指标中TC、LDL含量和LDL/HDL在高脂饲粮组显著提高(P<0.05),HDL和TGs含量在2组间没有显著差异(P>0.05)。

表2 鹌鹑血浆中脂肪代谢指标Table 2 The lipid plasma metabolism indices of Cotur nix japonica
2.2 盲肠微生物群落结构的比较
测序结果经过去噪过滤和质量控制后总共得到高质量数据257 860条,平均读长为545 bp,平均每个样品有(16 116±4 269)个序列。所有序列被归类到366种微生物(OTUs),平均每个样品有OTUs(123.00±24.75)个/组。方差分析结果(表3)显示高脂饲粮组和对照饲粮组之间序列数量没有显著差异(P>0.05),但是高脂饲粮组盲肠微生物OTUs显著减少(P<0.05)。

表3 测序数量及OTUs分析Table 3 Analysis of Sequencing quantities and OTUs
由图1可见,使用全部OTUs对微生物群落的β多样性进行PCA分析显示,高脂饲粮组与对照饲粮组被明显分成2组,这个结果说明2组的盲肠微生物群落结构和组成不同,PERMANOVA分析表明高脂饲粮组显著影响鹌鹑盲肠微生物群落的结构和组成(P<0.05)。
2.3 盲肠微生物群落在门、科和属分类水平的比较
为了寻找2组间微生物群落结构和组成的不同,我们对微生物不同分类单位进行比较研究。由图2可见,对序列数据进行匹配归类显示366个OTUs归属于6大门,序列主要分布于Firmicutes (77%)和Bacteroidetes(21%),其他序列属于如Spirochaetes、Tenericutes、Proteobacteria和Acti⁃nobacteria,总共占序列的2%。方差分析结果显示,序列分布在6个门的水平,高脂饲粮组和对照饲粮组之间没有显著差异(P>0.05)。微生物分类单位越大高脂饲粮的影响越深。在科的水平,高脂饲粮显著降低Lactobacillales、Streptococcaceae 和Clostridiales;其他(others)的序列数(P<0.05),显著提高Erysipelotrichales、Erysipelotrichaceae的序列数(P<0.05)(表4)。如图3所示,在科的水平,虽然统计分析差异不限制,高脂饲粮组的鹌鹑盲肠微生物还主要包括Clostridiaceae科,而对照组鹌鹑盲肠微生物还主要有Lactobacillaceae科。进一步分析发现在微生物属水平,高脂饲粮显著降低Ruminococcaceae、Ruminococcus的丰度(P<0.05),但是显著提高了Erysipelotrichaceae、cc_115和Erysipelotrichaceae、Eubacterium的丰度(P<0.05)(表5)。
2.4 盲肠微生物丰度与血浆脂肪代谢指标的相关性
盲肠微生物丰度与血浆脂肪代谢指标的相关性分析如图5所示,cc_115(Erysipelotrichaceae)和未被分类的微生物属于Lachnospiraceae科的丰度与TC、LDL含量和LDL/HDL呈显著正相关(P<0.05),除此之外Eubacterium(Erysipelotrichace⁃ae)只与LDL/HDL呈显著正相关(P<0.05)。Ru⁃minococcus(Ruminococcaceae)的丰度与TC、LDL含量和LDL/HDL呈显著负相关(P<0.05)。Ru⁃minococcus(Lachnospiraceae)的丰度与HDL含量呈显著正相关(P<0.05)。
3 讨 论
对微生物进行分类发现,日本鹌鹑盲肠微生物的组成在不同的分类单元与人类、小鼠、仓鼠、鸡和美洲鹌鹑体内的微生物相似[18-22]。在微生物门的水平,Bacteroidetes和Firmicutes是日本鹌鹑盲肠微生物的主要组成,超过98%的序列都属于这两大门类。在科的水平,主要包括Rikenellace⁃ae、Lactobacillaceae、Streptococcaceae、Lachnospir⁃aceae、Coprobacillaceae和Erysipelotrichaceae,在属的水平,微生物主要包括Ruminococcus、Blautia、Coprococcus和Eubacterium。
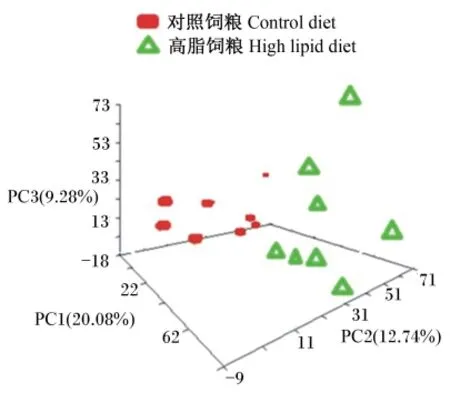
图1 盲肠微生物群落主成分分析三维图Fig.1 Three⁃dimensional projection of PCA of cecal microbial community
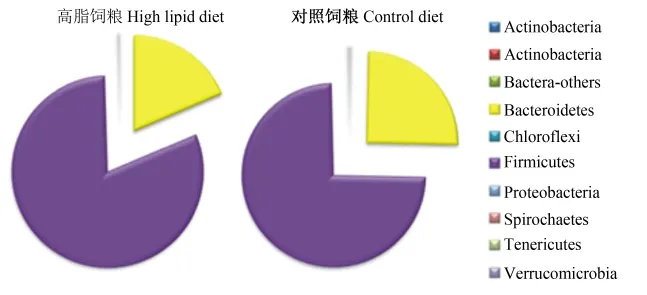
图2 盲肠微生物在门水平分布Fig.2 The pie chart of cecal microbial abundance on phyla level
研究报道降低肠道微生物的多样性会对宿主免疫产生影响,进而影响健康,产生代谢疾病[23-24]。本研究饲喂高脂饲粮后,测序数据量2组之间没有差异,但是鹌鹑盲肠OTUs显著降低,说明盲肠微生物的种类多样性显著减少。PCA分析显示高胆固醇饲粮组与对照组盲肠微生物群落结构和组成不同,高胆固醇饲粮显著影响了各个分类单元微生物的丰富度,改变了盲肠微生物群落结构和组成。研究发现饲喂老鼠高脂饲粮增加了Bacteroidetes丰度和减少了Firmicutes丰度,其他研究也有类似发现[25-26]。我们发现饲喂鹌鹑高脂饲粮,盲肠微生物Bacteroidetes有增加趋势和Firmicutes存在减少趋势,但是没有生物统计上的差异,导致这样的结果可能是每个组有8个样品,样品量不够达到显著性,所以今后的研究中需要增加采样量验证。进一步分析发现高脂饲粮影响微生物大的分类单位。在科的水平,高脂饲粮显著降低Firmicutes门中Lactobacillales、Streptococ⁃caceae和Clostridiales、others的丰度,但是显著提高Erysipelotrichales、Erysipelotrichaceae的丰度。显著增加的微生物引起我们的注意,研究表明人和动物肠道中Erysipelotrichaceae科微生物能产生短链脂肪酸(SCFAs)[27],主要包括乙酸盐、丙酸盐和丁酸盐[28]。大部分SCFAs是能量来源,能为人体提供10%的日常能量[29],为猪供应30%的能量[30],70%的乙酸盐被肝脏利用[31]。所以对于显著增加的Erysipelotrichaceae科微生物,我们推测该科微生物与动物体能量代谢相关,进一步研究发现Erysipelotrichaceae科中的cc_115和Eubacte⁃rium属丰度显著增加。通过微生物丰度与血浆脂肪代谢指标的相关性分析发现,Erysipelotrichaceae科中cc_115属微生物丰度与TC、LDL含量和LDL/HDL呈显著正相关。在本次研究还发现,虽然在统计分析中不显著,但是高脂饲粮减少Lacto⁃bacillaceae科的丰富度,前人研究表明高脂饲粮显著减少该科微生物,与引起宿主的低炎症反应,增加血浆中内毒素相关[32-34],饲喂高脂饲粮的鹌鹑机体是否存在低炎症,需要进一步的试验验证。肠道微生物的活动作为一个整体,受到干扰或者病态的群落结构和组成会使某些微生物过度生长或者减少,所以打破平衡的微生物群落会对宿主健康产生一定影响。
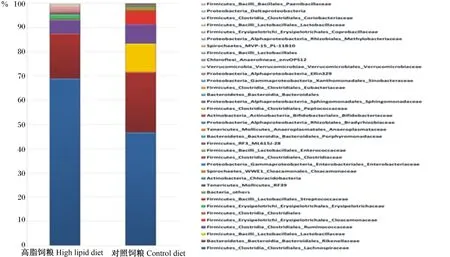
图3 盲肠微生物在科水平分布Fig.3 The histogram of cecal microbial abundance on family level

表4 盲肠微生物在科水平显著性分析Table 4 Family level significance analysis of cecal microbial 条/组
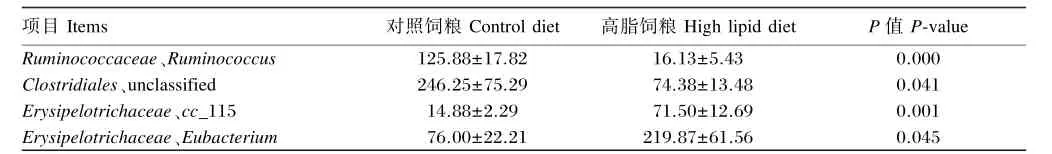
表5 盲肠微生物在属水平显著性分析Table 5 Genus level significance analysis of cecal microbial 条/组
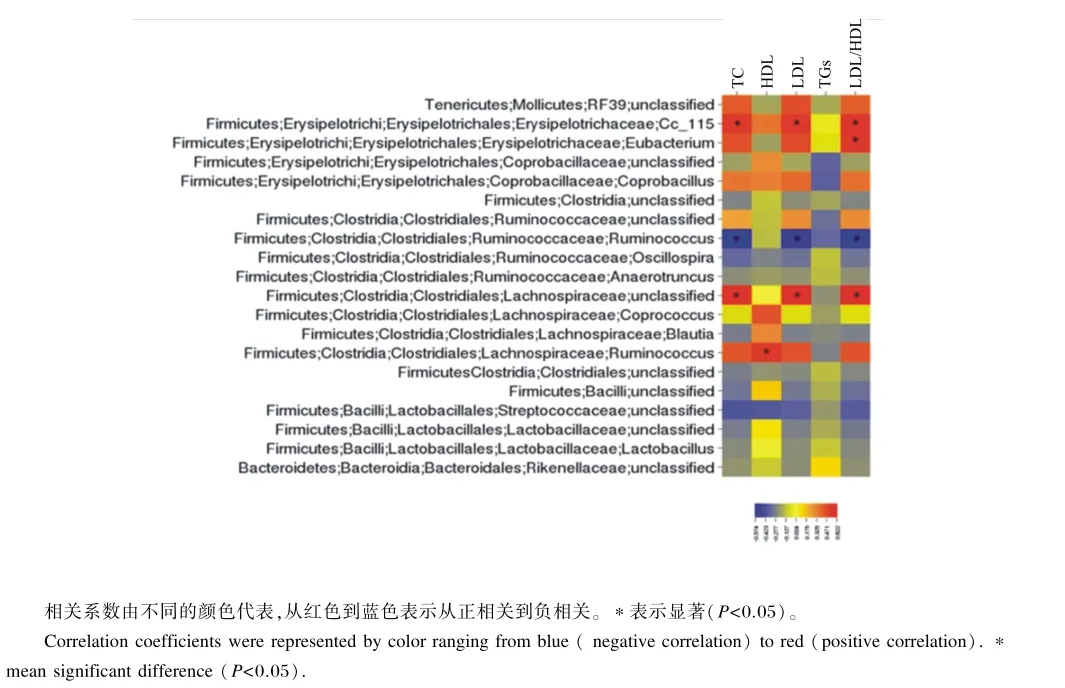
图4 盲肠微生物在属水平丰富度与脂肪代谢指标相关性分析热图Fig.4 Correlations heat map demonstrating the association between the abundances of different cecal microbial genera and plasma lipid parameters
本试验中,高脂饲粮组鹌鹑血浆中TC、LDL含量和LDL/HDL显著高于对照组,而且盲肠中一些微生物的丰度与脂肪代谢存在相关性。cc_115 (Erysipelotrichaceae)和未被分类的微生物属于Lachnospiraceae科与TC、LDL含量和LDL/HDL呈显著正相关,有报道指出人的粪便中属于Erys⁃ipelotrichaceae和Lachnospiraceae科的微生物丰度与这3个脂肪代谢指标存在显著正相关[18]。除此之外,鹌鹑研究还发现Eubacterium(Erysipe⁃lotrichaceae)只与LDL/HDL呈显著正相关。Ru⁃minococcus(Ruminococcaceae)的丰度与TC、LDL含量和LDL/HDL呈显著负相关。Ruminococcus (Lachnospiraceae)的丰度与HDL含量呈显著正相关。这个信息为今后进一步研究具体细菌株与能量代谢的相关性提供了信息。
4 结 论
使用焦磷酸测序技术发现了高脂饲粮能显著显著降低日本鹌鹑盲肠微生物的多样性,Firmi⁃cutes门中Lactobacillales;Streptococcaceae和Clos⁃tridiales;others的丰度,但是显著提高Erysipe⁃lotrichales;Erysipelotrichaceae的丰度,且Erysipe⁃lotrichaceae科中cc_115属微生物丰度与能量代谢有相关性。
参考文献:
[1] ZHAO L,WANG G,SIEGEL P,et al.Quantitative ge⁃netic background of the host influences gut microbi⁃omes in chickens[J].Scientific Reports,2013,3:1163.
[2] WHITMAN W B,COLEMAN D C,WIEBE W J.Pro⁃karyotes:the unseen majority[J].Proceedings of the National Academy of Sciences of the United States of America,1998,95(12):6578-6583.
[3] BÄCKHED F,DING H,WANG T,et al.The gut mi⁃crobiota as an environmental factor that regulates fat storage[J].Proceedings of the National Academy ofSciences of the United States of America,2004,101 (44):15718-15723.
[4] BÄCKHED F,MANCHESTER J K,SEMENKOV⁃ICH C F,et al.Mechanisms underlying the resistance to diet⁃induced obesity in germ⁃free mice[J].Proceed⁃ings of the National Academy of Sciences of the Unit⁃ed States of America,2007,104(3):979-984.
[5] SEKIROV I,RUSSELL S L,ANTUNES L C M,et al.Gut microbiota in health and disease[J].Physiological Reviews,2010,90(3):859-904.
[6] LOZUPONE C A,STOMBAUGH J I,GORDON J I,et al.Diversity,stability and resilience of the human gut microbiota[J].Nature,2012,489(7415):220-230.
[7] GOOTJES J,TEL R M,BERGKAMP F J M,et al.La⁃boratory evaluation of a novel capillary blood sam⁃pling device for measuring eight clinical chemistry pa⁃rameters and HbA1c[J].Clinica Chimica Acta,2009,401(1/2):152-157.
[8] WARNICK G R,ALBERS J.A comprehensive evalu⁃ation of the heparin⁃manganese precipitation procedure for estimating high density lipoprotein cholesterol[J].Journal of Lipid Research,1978,19(1):65-76.
[9] WARNICK G R,NAUCK M,RIFAI N.Evolution of methods for measurement of HDL⁃cholesterol:from ultracentrifugation to homogeneous assays[J].Clinical Chemistry,2001,47(9):1579-1596.
[10] FRIEDEWALD W T,LEVY R I,FREDRICKSON D S.Estimation of the concentration of low⁃density lipo⁃protein cholesterol in plasma,without use of the pre⁃parative ultracentrifuge[J].Clinical Chemistry,1972,18(6):499-502.
[11] OKADA M,MATSUI H,ITO Y,et al.Low⁃density lipoprotein cholesterol can be chemically measured:a new superior method[J].Journal of Laboratory and Clinical Medicine,1998,132(3):195-201.
[12] CAPORASO J G,KUCZYNSKI J,STOMBAUGH J,et al.QIIME allows analysis of high⁃throughput com⁃munity sequencing data[J].Nature Methods,2010,7 (5):335-336.
[13] QUINCE C,LANZEN A,DAVENPORT R J,et al.Removing noise from pyrosequenced amplicons[J].BMC Bioinformatics,2011,12(1):38.
[14] WANG Q,GARRITY G M,TIEDE J M,et al.Naive Bayesian classifier for rapid assignment of rRNA se⁃ quences into the new bacterial taxonomy[J].Applied and Environmental Microbiology,2007,73(16):5261-5267.
[15] CAPORASO J G,BITTINGER K,BUSHMAN F D,et al.PyNAST:a flexible tool for aligning sequences to a template alignment[J].Bioinformatics,2010,26 (2):266-267.
[16] ANDERSON M J,GORLEY R N,CLARKE K R.Permanova+for primer:guide to software and statisticl methods[M].Plymouth:Plymouth Marine Laborato⁃ry,2008.
[17] MCCUNE B,MEFFORD M J.PC⁃ORD:multivariate analysis of ecological data:MjM Software Design [Z].1999.
[18] KARLSSON F H,FÅK F,NOOKAEW I,et al.Symp⁃tomatic atherosclerosis is associated with an altered gut metagenome[J].Nature Communications,2012,3 (12):1245.
[19] TURNBAUGH P J,BÄCKHED F,FULTON L,et al.Diet⁃induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome[J].Cell Host&Microbe,2008,3(4):213-223.
[20] VIDENSKA P,RAHMAN M M,FALDYNOVA M,et al.Characterization of egg laying hen and broiler fe⁃cal microbiota in poultry farms in Croatia,Czech Re⁃public,Hungary and Slovenia[J].PLoS One,2014,9 (10):e110076.
[21] SU H W,MCKELVEY J,ROLLINS D,et al.Cultiva⁃ble bacterial microbiota of northern Bobwhite(Coli⁃nus virginianus):a new reservoir of antimicrobial re⁃sistance?[J].PLoS One,2014,9(6):e99826.
[22] BENNETT D C,TUN H M,KIM J E,et al.Character⁃ization of cecal microbiota of the emu(Dromaius no⁃vaehollandiae)[J].Veterinary Microbiology,2013,166(1/2):304-310.
[23] TURNBAUGH P J,HAMADY M,YATSUNENKO T,et al.A core gut microbiome in obese and lean twins[J].Nature,2009,457(7228):480-484.
[24] HILDEBRAND F,NGUYEN T L A,BRINKMAN B,et al.Inflammation⁃associated enterotypes,host geno⁃type,cage and inter⁃individual effects drive gut micro⁃biota variation in common laboratory mice[J].Ge⁃nome Biology,2013,14(1):R4.
[25] CANI P D,BIBILONI R,KNAUF C,et al.Changes in gut microbiota control metabolic endotoxemia⁃inducedinflammation in high⁃fat diet⁃induced obesity and dia⁃betes in mice[J].Diabetes,2008,57(6):1470-1481.
[26] DANIEL H,GHOLAMI A M,BERRY D,et al.High⁃fat diet alters gut microbiota physiology in mice[J].The ISME Journal,2014,8(2):295-308.
[27] JUMPERTZ R,LE D S,TURNBAUGH P J,et al.En⁃ergy⁃balance studies reveal associations between gut microbes,caloric load,and nutrient absorption in hu⁃mans[J].The American Journal of Clinical Nutrition,2011,94(1):58-65.
[28] WONG J M,DE SOUZA R,KENDAL C W,et al.Colonic health:fermentation and short chain fatty acids [J].Journal of Clinical Gastroenterology,2006,40 (3):235-243.
[29] ZAPOLSKA⁃DOWNAR D,NARUSZEWICZ M.Pro⁃pionate reduces the cytokine⁃induced VCAM⁃1 and ICAM⁃1 expression by inhibiting nuclear factor⁃kappa B(NF⁃kappaB)activation[J].Journal of Physiology and Pharmacology,2009,60(2):123-131.
[30] VON HEIMENDAHL E,BREVES G,ABEL H.Fiber⁃ related digestive processes in three different breeds of pigs[J].Journal of Animal Science,2010,88(3):972-981.
[31] BLOEMEN J G,VENEMA K,VAN DE POLL M C,et al.Short chain fatty acids exchange across the gut and liver in humans measured at surgery[J].Clinical Nutrition,2009,28(6):657-661.
[32] CANI P D,NEYRINCK A M,FAVA F,et al.Selec⁃tive increases of bifidobacteria in gut microflora im⁃prove high⁃fat⁃diet⁃induced diabetes in mice through a mechanism associated with endotoxaemia[J].Diabeto⁃logia,2007,50(11):2374-2383.
[33] CANI P D,AMAR J,IGLESIAS M A,et al.Metabolic endotoxemia initiates obesity and insulin resistance [J].Diabetes,2007,56(7):1761-1772.
(责任编辑 武海龙)
[34] KIM K A,GU W,LEE I A,et al.High fat diet⁃in⁃duced gut microbiota exacerbates inflammation and o⁃besity in mice via the TLR4 signaling pathway[J].PLloS One,2012,7(10):e47713.
Effects of High Lipid Diet on Cecal Microbial Community Structure of Japanese Quail(Cotur nix japonica)
LIU Shasha1,2ZHANG Hongfu1∗ZHEMG Mingde3JI Cheng2
(1.State Key Laboratory of Animal Nutrition,Institute of Animal Sciences,Chinese Academy of Agricultural Sciences,Beijing 100193,China;2.College of Animal Science,Chinese Agruculture University,Beijing 100193,China;3.Avian Research Centre,Faculty of Land and Food Systems,The University of British Columbia,Vancouver V6T1Z4,Canada)
∗Corresponding author,professor,E⁃mail:zhanghf656@vip.sina.com
Abstract:The object of this study was to investigate the effects of high lipid diet on cecal microbial community structure and composition and its lipid metabolism of Japanese quail(Cotur nix japonica).A total of 160 one⁃day⁃old Japanese quail fed a semi⁃synthetic diet for the first 6 weeks.At 6 weeks of age,the birds were ran⁃domly divided into 2 groups with 8 replicates per group and 10 birds per replicate,fed either a regular synthetic diet or a synthetic diet with added cholesterol for another 6 weeks.After the feeding trial,8 birds from each group were euthanized and collected blood for testing the lipid metabolism indices,cecal contents for investiga⁃ting the microbial community by using pyrosequencing.The results showed as follows:1)The content of total cholesterol(TC),low density lipoprotein(LDL)and LDL/high density lipoprotein(HDL)of Japanese quail were significantly increased in high lipid diet group(P<0.05).2)High lipid diet significantly decreased cecal microbial diversity(P<0.05);in high lipid diet group,the abundance of family Lactobacillales,Streptococ⁃caceae and Clostridiales,others was significantly decreased(P<0.05),while the abundance of family Erysip⁃elotrichales,Erysipelotrichaceae was significantly increased(P<0.05).3)The abundance of cc_115(Erysip⁃elotrichaceae)and unclassified bacteria belong to Lachnospiraceae were positively correlated with contents of TC,LDL and LDL/HDL(P<0.05),besides Eubacterium(Erysipelotrichaceae)was only positively correla⁃ted with LDL/HDL(P<0.05),while the abundance of Ruminococcus(Ruminococcaceae)was negatively correlated with contents of TC,LDL and LDL/HDL(P<0.05),and the abundance of Ruminococcus(Lach⁃nospiraceae)was positively correlated with contents of HDL(P<0.05).In conclusion,high lipid diet highly affects the cecal microbial community structure and composition and its lipid metabolism of Japanese quail.[Chinese Journal of Animal Nutrition,2015,27(8):2368⁃2377]
Key words:high lipid diet;cecal microbial community;lipid metabolism
作者简介:刘莎莎(1985—),女,河南安阳人,博士研究生,动物营养与饲料专业。E⁃mail:King123apple@126.com
收稿日期:2015-02-16
doi:10.3969/j.issn.1006⁃267x.2015.08.008
文章编号:1006⁃267X(2015)08⁃2368⁃10
文献标识码:A
中图分类号:S816
通信作者:∗张宏福,研究员,博士生导师,E⁃mail:zhanghf656@vip.sina.com
