辅助量子化学计算的红外光谱课堂教学
2015-12-27陈广慧罗承源姜喜双
江 霞,陈广慧,罗承源,姜喜双
(1.汕头大学理学院,广东汕头 515063;2.辽宁工程技术大学电控学院,辽宁葫芦岛 125000)
辅助量子化学计算的红外光谱课堂教学
江 霞1,陈广慧1,罗承源1,姜喜双2
(1.汕头大学理学院,广东汕头 515063;2.辽宁工程技术大学电控学院,辽宁葫芦岛 125000)
传统的化学专业本科生红外光谱课堂教学仅凭黑板和粉笔,授课形式单一,无法展示有机分子中原子的振动模式,学生很难将红外光谱理论联系实验.文章分别采用ChemBioOffice和Gaussian 09量子化学计算程序包,在量子化学半经验和密度泛函理论水平对苯分子进行优化并作振动频率计算,通过可视化软件GaussView对苯分子的振动模式进行了归属,并采用VEDA4程序进一步分析苯分子红外吸收峰对应的振动模式分布.通过在《波谱分析》课堂教学中加以演示动态的振动模式,可以加深学生对分子红外光谱的理解.
红外光谱;振动模式;量子化学计算;ChemBioOffice;Gaussian 09
红外光谱(Infrared Spectroscopy,IR)的研究始于20世纪初,自1940年红外光谱仪问世,红外光谱法作为一种常规的仪器分析方法在化学实验工作中广泛应用.其原理是当一束具有连续波长的红外光通过物质,物质分子中某个基团的振动频率或转动频率和红外光的频率一样时,分子就吸收能量由原来的基态振(转)动能级跃迁到能量较高的振(转)动能级,分子吸收红外辐射后发生振动和转动能级的跃迁,该处波长的光就被物质吸收.所以,红外光谱法实质上是一种根据分子内部原子间的相对振动和分子转动等信息来确定物质分子结构和鉴别化合物的分析方法.
分子中原子振动谐振子本质属于微观粒子体系,描述其运动规律要采用量子力学薛定谔方程.简谐振动体系的势能为

求解体系能量的薛定谔方程

得
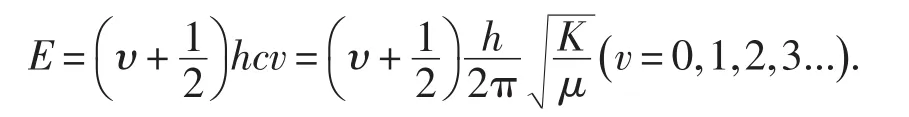
式中:v是振动量子数,E是与振动量子数v相对应的体系能量.
量子化学程序计算频率的过程大致可分为三个步骤:(1)优化电子结构,得到电子的Slater行列式;(2)Slater行列式为基础,求得能量对位移的二阶导数,即力常数矩阵;(3)对角化力常数矩阵,获得频率,数学表示式如下.设有一个经过全优化的分子体系,其力常数矩阵K为

在《波谱分析》课程中,一般采用传统的粉笔+黑板教学手段.由于红外光谱概念相对抽象,学生在学习时普遍感到难以理解,单凭分子结构和静态的红外谱图对分子振动模式的理解有很大困难,影响了教学效果.随着计算机技术的发展和量子化学计算的发展,越来越多的量子化学计算软件和化学作图分析软件应用于化学研究领域中,化学学科不再是一门纯粹的实验科学.例如,量子化学计算可以模拟红外光谱并展示红外振动模式[1,2].而将这种方法应用到课堂当中,能使抽象、枯燥的化学理论知识变得形象、具体,激发学生的学习热情.
本文尝试采用ChemBioOffice[3]在量子化学半经验和Gaussian 09[4]程序包在密度泛函理论(DFT)对苯分子进行结构优化和振动频率计算,并结合可视化软件GaussView[5,6]模拟苯分子红外光谱,对振动模式进行动态演示并作全面的归属.此外,采用VEDA4[7,8]程序分析苯的吸收峰对应的振动模式分布.通过展示有机分子的化学结构和这些谱图的关系,使学生深刻理解谱图产生的本质,进而学会采用量子化学计算方法预测红外谱图.
1 计算方法
采用ChemBioOffice分别在MNDO、AM1和PM3水平以及Gaussian 09程序包在B3LYP/6-31G(d)水平对苯分子结构进行优化,得到平衡几何结构.在此基础上采用相同的理论水平计算得到苯分子振动频率.
2 苯分子的平衡几何结构
分别在MNDO、AM1、PM3和B3LYP/6-31G(d)水平对苯分子进行优化,得到D6h分子点群的平衡结构.对此结构进行频率计算,没有发现虚频,说明优化的几何结构为稳定结构.优化后的苯分子几何参数如图1所示.MNDO水平优化的C-C和C-H键长分别为1.406和1.090Å;AM1水平优化得到的C-C和C-H键长分别为1.395和1.099Å;PM3水平优化的C-C和C-H键长分别为1.391和1.095Å;B3LYP/6-31G(d)水平得到的C-C和C-H键长分别为1.397和1.087Å.对比实验[9]测得苯分子的C-C和C-H键长1.400和1.080Å,发现B3LYP/6-31G(d)水平优化得到的几何结构更为接近,最大偏差仅为0.6%,表明该计算方法是相对可靠的.
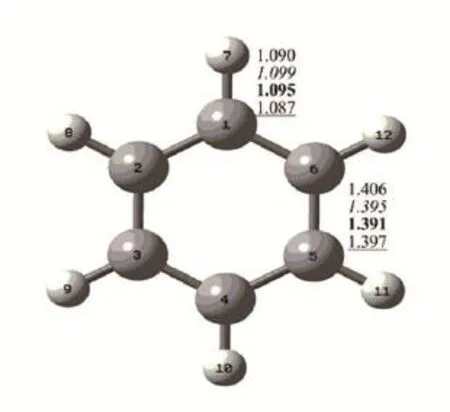
图1 MNDO、AM1(斜体)、PM3(加粗)和B3LYP/ 6-31G(d)(下划线)水平下优化的苯分子几何构型
3 苯分子振动频率的计算
在结构优化的基础上,使用相同理论水平计算苯分子的振动光谱.只有在振动中伴随偶极矩变化的振动模式才具有红外吸收,即具有红外活性.对于含有N个原子的非线性分子,分子的振动模式有3N-6个.苯分子有12个原子,对应有30个振动模式,其中只有4个具有红外活性.
3.1 采用ChemBioOffice计算
众所周知,ChemBioOffice是一个广泛应用的化学作图软件,除了能够直观、简单地描述分子结构和反应过程,还有一个常常被忽略的功能——其中的ChemBio3D可以预测化合物红外光谱.对于初学者使用ChemBio3D模拟红外光谱是个不错的选择,因为其使用比较简单.由于半经验方法计算速度快、所需的磁盘空间和计算机内存小、可计算的分子体系大,所以为了方便计算,首先采用ChemBio3D在半经验的理论水平下模拟苯的红外光谱.
从表1所示的计算的红外振动频率可以看出,采用MNDO方法得到苯分子C-H面外摇摆、两个C=C和一个C-H伸缩振动频率分别为767.5、1 146.1、1 555.0和3 436.5 cm-1;采用AM1方法得到苯分子C-H面外摇摆、两个C=C和一个C-H伸缩振动频率分别为743.8、1145.3、1 579.0和3 187.8 cm-1;采用PM3方法得到苯分子C-H面外摇摆、两个C=C和一个C-H伸缩振动频率分别为712.6、1 068.3、1 547.0和3 083.5 cm-1.对比实验值676.0、1 035.0、1 478.0和3 091.0 cm-1[9],发现PM3方法更为接近,最大偏差为5.4%.通过多次对不同化合物IR光谱的对比,发现ChemBio3D对于简单有机物的IR预测多数情况下还是基本准确的,但是其对于复杂的化合物仅能做一些粗糙的定性分析,而达不到精确计算的目的.ChemBio3D中半经验水平的Predict IR工具,作为教学用途中预测简单有机物的红外光谱已经足够.但是为了精确获得复杂体系的红外光谱,同时也为了提高学生利用量子化学计算程序模拟红外光谱的能力,有必要进行更高水平的理论研究.

表1 在MNDO、AM1、PM3和B3LYP/6-31G(d)水平得到苯分子红外振动频率以及相应实验值(单位:cm-1)
3.2 采用Gaussian 09程序包计算
利用Gaussian 09采用高水平量子化学方法预测小分子体系红外光谱(IR),后自洽场(post-HF)方法包括MP2、MP3、MP4、CCSD、CCSD(T)以及MC-SCF等,可以给出精确的红外光谱.但是其计算量过大、耗时,分别与电子数目(N)N5、N6成正比.相比之下,密度泛函理论(DFT)[10,11]同样也考虑了电子相关,计算结果同样可靠.但是其优势在于所需计算机时较少,计算时间与电子数目(N)N3成正比,所得分子结构和IR谱等与实验谱图一致性高,不论在化学反应还是物理性质研究方面都有广泛应用.特别值得注意,DFT方法是计算分子结构、研究分子性质中最常用的方法之一,是可与分子轨道理论相提并论的严格非波函数型量子化学计算理论[12].
在B3LYP/6-31G(d)水平计算苯分子振动频率,计算结果如表1所示.从表中数据可以看出,在该水平下得到苯分子C-H面外摇摆、两个C=C和一个C-H伸缩振动频率分别为691.8、1 069.3、1 531.6、3 203.8 cm-1,对比实验值676.0、1 035.0、1 478.0、3 091.0 cm-1[9],发现最大偏差仅为3.6%.可以看出在B3LYP/6-31G(d)水平下得到振动频率与实验值比较接近,说明其比半经验方法更准确.
理论计算与实验值存在一定的误差主要源于以下因素:理论计算的分子处于气态状态下,是单分子的行为.而实验中的IR检测是多分子的堆积形式.此外,也存在一定的方法和基组误差.所以理论计算值与实验值存在一定的差异是合理的.为了使量子化学计算模拟的红外光谱能够更接近实验值,人们通常在计算中引入校正因子[13].尝试用不同的校正因子来校正B3LYP/6-31G(d)水平下得到频率,当校正因子取0.965时,计算的频率与实验相比最大偏差仅为-1.2%,如表1所示.
4 苯分子红外谱图的模拟及振动模式分析
在红外光谱分析中,除了分子结构优化和振动频率计算以外,谱线的指认也是谱图解析的关键.对于复杂分子的化合物而言,谱线的正确归属对化合物分子结构的确定具有十分重要的意义.可以利用Gauss-View模拟苯分子红外谱图,动态演示其振动模式.此外,采用VEDA4程序进一步分析苯4个吸收峰对应振动模式的分布.
4.1 采用GaussView模拟苯红外谱图及分析其振动模式
用GaussView打开B3LYP/6-31G(d)水平优化苯分子的输出文件,通过Spectrum选项可以直接模拟如图2所示的红外光谱图.发现模拟谱图的分子基团的特征吸收峰与实验谱图一一对应,振动强度也很接近.通过Vibrations选项以动画的形式动态显示其红外特征吸收峰处的振动模式,如图3所示(原子的运动方向如箭头所示),很容易判断苯分子的4个红外特征吸收峰对应的振动模式:(a)C-H面外摇摆振动;(b)C=C伸缩振动;(c)C=C伸缩振动;(d)C-H伸缩振动.

图2 量子化学模拟与实验[9]测得的苯分子红外吸收光谱
4.2 采用VEDA4程序分析苯4个特征振动模式分布
采用可视化软件GaussView以动画的形式循环播放其特征吸收峰处的振动模式时,可以直观地看到,当某个化学键伸长或者缩短的时候,会同时带动与之相连的其他键的运动,也就是说在该简正振动模式中同时包含了一定成分的其他振动模式.由此可见,分子基团的振动都不是完全孤立的,而是离域化的,并且在一定程度上与其他官能团的振动运动相耦合.采用GaussView并不能精确判断每个吸收峰所对应的振动模式以及对该吸收峰的贡献,而采用VEDA4程序就可以做到.图1对应B3LYP/6-31G(d)水平下优化得到苯分子结构示意图,表2给出了该理论水平下苯的4个特征吸收峰对应的振动模式分布结果.例如苯分子在691.8 cm-1处的特征红外吸收峰是由二面角H7-C1-C6-C5、 H8-C2-C3-C4、 H9-C3-C4-C5、H10-C4-C5-C6、H11-C5-C6-C1和H12-C6-C5-C4的变形耦合而成的,它们对该吸收峰的贡献分别为17.0%、17.0%、16.0%、17.0%、17.0%和16.0%;在1069.3cm-1处的特征红外吸收峰是由C6-C5的键长伸缩、键角H7-C1-C6、H9-C3-C4、H10-C4-C5、H12-C6-C5、C5-C4-C3和C6-C5-C4的变形耦合而成的,它们对该吸收峰的贡献分别为14.2%、11.3%、12.3%、10.4%、10.4%、25.5%和16.0%.通过振动模式分布分析,学生能够定量地判断复杂分子中红外吸收峰的振动模式分布.这样不仅能够有效完成课堂教学,更能够为学生将来开展定性定量分析提供一个有效的途径.

图3 在B3LYP/6-31G(d)水平下计算的苯分子4个红外振动模式

表2 B3LYP/6-31G(d)水平下的苯分子4个吸收峰对应的振动模式分布
5 结论
本文系统地介绍了ChemBioOffice和Gaussian 09程序包模拟红外光谱(IR)的基本原理和应用.由于ChemBioOffice采用的是半经验方法,不能精确模拟所有化合物的红外光谱.但是作为在《波谱分析》课堂上的演示手段,其简便易行,可以帮助学生加深对波谱分析教科书中红外光谱理论的理解.对于复杂化合物红外光谱可以采用基于第一性原理的Gaussian 09程序包进行模拟,帮助学生掌握采用密度泛函理论预测光谱这一重要方法.此外,还采用GaussView对苯分子的振动模式进行动态演示并作全面的归属,通过在《波谱分析》课堂教学中加以演示动态的振动模式,可以加深学生对分子红外光谱的理解.利用VEDA4程序定量地判断复杂分子中红外吸收峰的振动模式分布,为学生将来开展定性、定量分析提供一个有效的途径.利用上述计算程序和可视化软件在《波谱分析》红外光谱学课堂进行辅助教学,将分子振动模式形象化,从而提供一种新的方法——量子化学计算方法预测红外谱图.能够有效提高学生的学习兴趣和积极性,提升教学质量.辅助量子化学计算的波谱分析教学改革,借助多种计算机软件将枯燥难懂的理论知识和微观的原子分子振动通过量子化学软件展现,使课堂教学直观、生动,使学生易于接受.
[1]唐敖庆,杨忠志,李前树.量子化学[M].北京:科学出版社,1982.
[2]徐光宪,黎乐民,王德民.量子化学-基本原理和从头算法[M].北京:科学出版社,1985.
[3]任萃毅.介绍一个新型的化学软件包-CS Chem Office 2000[J].化学教学,2001(3):34-36.
[4]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09,Revision A.02.Gaussia n,Inc,Wallingford,CT.2010.
[5]LUNDBERG M,KAWATSU T,FRISCH M J,et al.DFT-ONIOM Study of the dopamine:β CD complex:NBO and AIM analysis[J].Canadian Journal of Chemistry,2015,93(10):1115-1121.
[6]DEVLIN F J,STEPHENS P J,OESTERLE C,et al.Configurational and conformational analysis of chiral molecules using IR and VCD spectroscopies:spiropentylcarboxylic acid methyl ester and spiropentyl acetate[J].The Journal of organic chemistry, 2002,67(23):8090-8096.
[7]SADLEJ J,DOBROWOLSKI J C,RODE J E,et al.DFT study of vibrational circular dichroism spectra of D-lactic acid-water complexes[J].Physical Chemistry Chemical Physics,2006,8(1):101-113.
[8]JAMRÓZ M H.Vibrational energy distribution analysis(VEDA):scopes and limitations,spectrochim[J].Spectrochimica Acta Part A:Molecular and Biomolecular Spectroscopy,2013,114:220-230.
[9]NIST Chemistry Webbook[EB/OL].(2015-10-16)[2012-11-06].http://webbook.nist.gov/chemistry.
[10]BECKE A D.Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior[J].Phys.Rev.A, 1988,38(6):3098-3100.
[11]YANG G,ZHOU L J,LIU X C,et al.Density Functional Calculations on the Distribution,Acidity,and Catalsis of TiIV and TiIIIIons in MCM-22 Zeolite[J].Chem.-A Euro.J,2011,17(5):1614-1621.
[12]杨玉琼.离域效应的量子力学处理[J].毕节师专学报,1995,1(2):76-78.
[13]SCOTT A C,RADOM L.Harmonic Vibrational Frequencies:An Evaluation of Hartree-Fock,Møller-Plesset,Quadratic Config-uration Interaction,Density Functional Theory,and Semiempirical Scale Factors[J].J.Phys.Chem,1996,100(41): 16502-16513.
The Teaching Method of Infrared Spectroscopy with the Auxiliary Quantum Chemistry Calculation
JIANG Xia1,CHEN Guang-hui1,LUO Cheng-yuan1,JIANG Xi-shuang2
(1.College of Science,Shantou University,Shantou,Guangdong,515063;2.College of Electronic Control,Liaoning Technical University,Huludao,Liaoning,125000)
The traditional infrared spectrum classroom teaching of undergraduates in chemistry is only conducted by the blackboard and the chalk,which cannot display the vibrational modes of the organic molecules.It is difficult for the students to contact experiment from infrared spectrum theory.In this article,optimization and vibration frequency calculation of benzene molecules were performed using the ChemBioOffice at semi-empirical theoretical level and Gaussian 09 program package at B3LYP/6-31G(d)theoretical level.The vibrational frequencies of benzene molecules were analyzed by using GaussView software,and the distribution of vibrational modes corresponding to absorption peaks of benzene were analyzed by VEDA4 program.By demonstrating the dynamic vibration modes in class teaching of“Spectrum Analysis”course can help students understand the infrared spectrum.
Infrared spectrum;vibrational modes;quantum chemistry calculation;ChemBioOffice;Gaussian 09
O 64
A
1007-6883(2015)06-0085-06
责任编辑 朱本华
2015-11-08
广东省高等学校教学质量与教学改革工程项目(项目编号:2013-2015);广东省研究生示范课程建设项目(项目编号:2015-2017);汕头大学本科教学改革研究项目(项目编号:2013-2015).
江霞(1990-),女,湖北广水人,汕头大学理学院在读硕士研究生.陈广慧教授为通讯作者.
