超高效液相色谱-四极杆-飞行时间质谱法同时测定猪肉中多类兽药残留
2015-12-26朱万燕徐文远许美玲
朱万燕 , 张 欣, 杨 娟, 徐文远, 许美玲
(临沂出入境检验检疫局检验检疫技术中心,山东 临沂276034)
由于各类兽药的理化性质存在较大差异,现有的兽药残留分析主要以分类检测为主[1,2];这些方法所能同时分析的药物种类较少,若要检测多类兽药,需进行多次的样品前处理,检测周期长,成本高,效率低,不能满足目前兽药种类不断增加、分析通量不断提高的需求。因此兽药多残留同时检测技术的开发是研究的重点和难点[3-7]。开发动物源性食品中多类兽药残留同时分析的方法十分必要。
高效液相色谱-四极杆-飞行时间质谱(HPLCQ-TOF MS)分辨率高,可对化合物进行精确分子质量的测定,可以精确测定小数点后4 位有效数字,并依据所测得的化合物的精确分子质量对其进行确证分析;同时在碰撞诱导解离(CID)模式下,得到化合物碎片离子的分子式和精确分子质量,可进一步对化合物的结构和裂解规律加以确证,定性准确性很高[8]。刘畅等[9]建立了猪肉中31 种β-受体激动剂的HPLC-Q-TOF MS 同时测定方法;彭丽等[10]建立了超高效液相色谱-四极杆-飞行时间质谱(UPLCQ-TOF MS)筛查牛奶中55 种药物的方法;高馥碟等[11]建立了UPLC-Q-TOF MS 快速筛查牛奶中13种农药和29 种兽药残留的方法。目前,有关猪肉中6 类(磺胺类、喹诺酮类、四环素类、硝基咪唑类、大环内酯类、β-内酰胺类)33 种兽药残留的UPLC-QTOF MS 方法鲜有报道。
QuEChERS 方法[12]是固相萃取技术(SPE)和基质固相分散技术(MSPD)的衍生和进一步发展,该方法快速、环保、经济,目前主要用于果蔬中农药残留的分析,在动物性食品检测中的应用较少[13,14]。
本文在确定的仪器条件下,建立了6 类33 种兽药基于精确质量数、保留时间和碎片离子信息的筛查数据谱库(每种兽药有在4 个不同碰撞能量下得到的碎片离子信息),并结合本文建立的QuECh-ERS/UPLC-Q-TOF MS 分析方法,实现了在无标准品的情况下对猪肉样品中33 种兽药进行高选择性的同时快速筛查和确认,还可以对检测到的目标化合物进行准确定量。
1 实验部分
1.1 仪器与试剂
Agilent 1290-6530 超高效液相色谱-四极杆-飞行时间质谱仪(美国Agilent 公司);CR22GⅢ高速冷冻离心机(日本HITACHI 公司);ULTRA-TURRAX T25 型均质器、GENIUS 3 基本型涡流混匀器、HS260 型调速多用振荡器(德国IKA 公司);R-210型旋转蒸发仪(瑞士Buchi 公司)。
标准物质(纯度:92.2% ~99.5%)均购自德国Dr. Ehrenstorfer 公 司;乙 腈(ACN)、甲 醇(MeOH)、乙酸乙酯(EtAc)、甲酸和乙酸(HAc)为色谱纯,均购自美国Tedia 公司;硫酸镁、乙二胺四乙酸二钠(Na2EDTA)、硫酸钠(Na2SO4)和氯化钠(NaCl)为分析纯,均购自国药集团化学试剂有限公司;C18、PSA(硅胶表面键合N-丙基乙二胺)、NH2、中性Al2O3、PAX(阴离子交换混合机理水可浸润型聚合物基质)、PCX(阳离子交换混合机理的水可浸润型聚合物)、PEP(硅胶表面键合聚苯乙烯/二乙烯苯,表面同时具有亲水性和憎水性基团)、GCB(石墨化炭黑)、弗罗里硅土吸附剂均购于天津Bonna-Agela 公司;实验用水为Millipore 纯水系统(美国Millipore 公司)制得的高纯水。
1.2 标准溶液配制
分别准确称取相当于25 mg 目标分析物的标准品于50 mL 烧杯中,用甲醇溶解并定量于25 mL容量瓶中,即得1 g/L 的标准储备液,-18 ℃下避光保存。实验中用空白样品提取液逐级稀释,根据每种目标化合物的定量限配制成不同浓度的系列基质混合标准工作液,现用现配。
1.3 样品前处理
提取:称取4.0 g(精确至0.01 g)肉类样品于50 mL 离心管中,加入20 mL 5% (v/v)乙酸乙腈溶液和0.1 g Na2EDTA,同时加入4.0 g 无水硫酸钠和2.0 g 氯化钠均质提取1 min,于9 000 r/min 下离心5 min,取上清液待净化。
净化:将上清液移入装有300 mg C18 和400 mg NH2吸附剂的50 mL 具塞离心管中,使吸附剂和提取液充分接触,振荡5 min,以9 000 r/min 冷冻(-2 ℃)离心5 min,静置10 min 后,取10 mL 上清液(相当于2.0 g 样品)于50 mL 鸡心瓶中,40 ℃旋转蒸发至近干,氮吹吹干。加入1 mL 0.1% (v/v)甲酸水-乙腈(9 ∶1,v/v)的定容液,涡旋混匀,经0.22 μm 滤膜过滤,滤液供UPLC-Q-TOF MS 测定。
1.4 仪器工作条件
UPLC 条件:ZORBAX SB-C18 色谱柱(100 mm×2.1 mm,3.5 μm)。流动相A 为乙腈,B 为0.1%(v/v)甲酸水溶液(含5 mmol/L 乙酸铵);流速0.4 mL/min。梯度洗脱程序:0 ~10 min,1% A ~30%A;10 ~12 min,30% A ~40% A;12 ~14 min,40%A;14 ~14.1 min,40% A ~100% A;14.1 ~22 min,100% A;22 ~22.1 min,100% A ~1% A。柱温40 ℃;进样量10 μL。
Q-TOF MS 条件:电喷雾离子(ESI)源,正离子模式;毛细管电压4 000 V;雾化气压力2.4×105Pa;干燥气温度325 ℃;干燥气流速10 L/min;鞘气温度325 ℃;鞘气流速11 L/min;参比离子为m/z 121.050 9 和922.009 8,用于实时校正;数据采集采用棒状图数据采集模式,扫描范围为m/z 50 ~1 700,扫描速率为3 spectra/s;产物离子数据在Target MS/MS 模式下设置固定的保留时间、母离子、碰撞能量等信息进行采集,数据采集与处理使用Agilent MassHunter Workstation Software(version B.04.00),包括数据采集、定性分析、定量分析软件。
2 结果与讨论
2.1 色谱条件的选择
[7],选取0.1% (v/v)甲酸水(含5 mmol/L 的乙酸铵)和乙腈作为流动相。本文所分析的33 种兽药以极性化合物为主,色谱峰出峰相对集中且出峰时间较早,对于前10 min 的梯度洗脱程序,需要缓慢增加乙腈的比例,保证极性化合物的分离;10 min 之后,只有少量化合物出现在这一区域,此阶段快速提高乙腈的比例可快速洗脱目标化合物,缩短检测时间。经过流动相梯度配比优化使得不同种类的兽药得到分离。目标化合物混合标准溶液的总离子流图(TIC)见图1。

图1 33 种兽药混合标准溶液的总离子流图Fig.1 Total ion chromatogram of a mixed standard solution of the 33 veterinary drugs
2.2 数据库的建立
配制1 000 μg/L 的标准溶液,在LC-Q-TOF MS 全扫描模式下进行测定,记录化合物的精确相对分子质量和保留时间;在Targeted MS/MS 采集界面输入目标物的母离子、保留时间和相应的碰撞能量,对目标物进行数据采集,选择碎片离子信息较为丰富的4 个碰撞能量下的质谱图,并导出CEF 文件,将CEF 文件导入PCDL 软件中,与对应的兽药信息相关联并保存,即得数据谱库。表1 列出了33种化合物的分子式、理论相对分子质量、保留时间和特征离子。

表1 33 种兽药的保留时间、理论相对分子质量和质谱参数Table 1 Retention times,theoretical relative molecular masses (Mr)and MS parameters for the 33 veterinary drugs
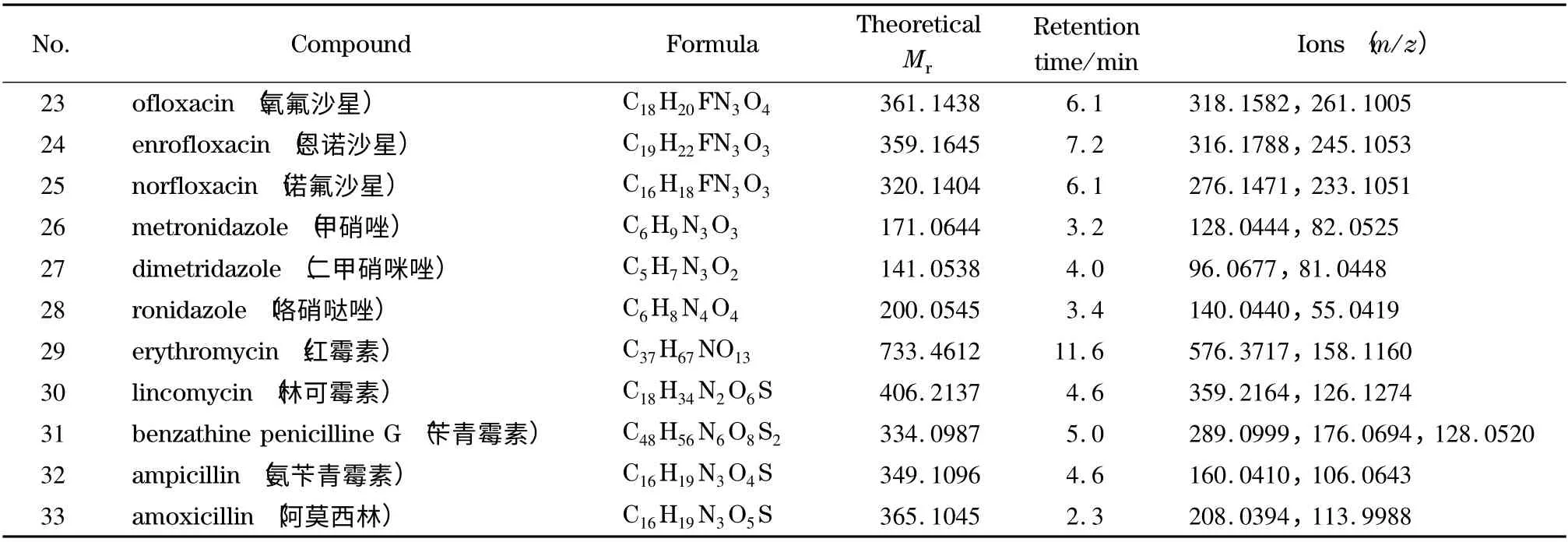
表1 (续)Table 1 (Continued)
根据欧盟2002/657/EC 决议要求,当使用高分辨质谱作为确证技术时,检测到1 个具有精确质量数的母离子就获得2 个确证分数,二级质谱中的每个子离子获得2.5 个确证分数,即1 个具有精确质量数的母离子和1 个特征碎片离子便可获得超过4分的确证分数。经1.3 节样品前处理后,结合本文建立的数据库,33 种兽药化合物均能够被四极杆-飞行时间质谱有效筛查出来。
2.3 检索参数的优化
筛查参数包括质量偏差窗口、保留时间窗口等。合适的参数设置可以避免假阳性和假阴性结果,提高筛查方法的准确性。
质量偏差窗口的确定:在LOQ、2LOQ 和4LOQ 3 个添加水平下,对样品中目标化合物的精确质量数偏差进行了考察。结果发现,84.8% 的目标化合物的质量偏差低于5×10-6(5 ppm),所有化合物的质量偏差均低于10×10-6(10 ppm)(见图2)。
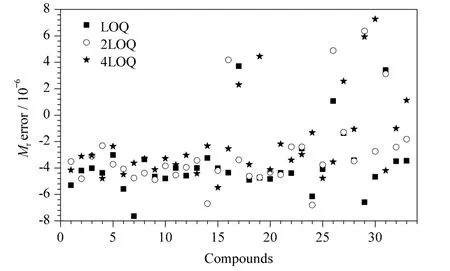
图2 33 种兽药在3 个添加水平下的质量偏差分布Fig.2 Distributions of Mr error for the 33 veterinary drugs at three spiked levels
保留时间窗口的确定:在样品基质中进行保留时间限定参数的评估,保留时间的限定范围分别为0.10、0.25、0.50、1.00 min 和无保留时间限定。实验结果表明,在保留时间限定为0.10、1.00 min 和无保留时间限定的条件下,待测化合物出现定性偏差的占比分别为8.3%、2.5% 和16.7%;保留时间限定为0.25、0.50 min 时,所有待测化合物均未出现定性偏差;此外,实验通过目标化合物保留时间的重复性(n =6)和重现性(n =6),对保留时间的稳定性进行了考察,结果表明日内保留时间最大标准偏差低于0.14 min,日间保留时间最大标准偏差低于0.24 min。最终保留时间的限定参数被设置为0.25 min。
2.4 前处理条件的优化
2.4.1 盐析剂和吸水剂的选择
对于水果蔬菜样品,QuEChERS 前处理普遍采用NaCl 作为盐析剂,MgSO4作为吸水剂。由于本方法没有添加水,NaCl 在本方法中的作用主要是辅助细胞破碎和目标物溶出,将样品中的少量水分析出,从而有利于吸水剂吸附。在QuEChERS 方法中添加MgSO4,主要是由于MgSO4能够吸收大量的水分。实验表明MgSO4作为吸水剂会明显降低部分兽药如磺胺类、喹诺酮类及四环素类的回收率,特别是四环素类药物。这是由于MgSO4中的Mg2+和四环素类兽药结合形成配合物,从而影响了四环素类兽药的提取,因此,实验选择Na2SO4替代MgSO4作为吸水剂。实验进一步考察了NaCl 和Na2SO4的用量,当添加2 g NaCl 和4 g Na2SO4时,两相分离效果最好,获得了较高的回收率。此外,为了抑制四环素类兽药与阳离子的配合作用,在提取液中加入0.1 g Na2EDTA。
2.4.2 提取剂的优化
实验对兽药残留检测常用提取试剂(乙腈、5%(v/v)乙酸乙腈、乙酸乙酯、甲醇)的提取效率进行了比较。从实验现象看,用乙腈、乙酸乙酯和乙酸乙腈提取时,提取液澄清;用甲醇提取时,提取液浑浊,这可能是由于甲醇与蛋白质形成絮状沉淀的原因,不利于后续净化和测定步骤的进行。从实验结果看,乙酸乙腈的提取效率最高,75.7% 的目标化合物的回收率在70% ~120% 范围内,且采用乙酸乙腈提取时,共提物较少,初步确定采用乙酸乙腈作为提取液。不同提取剂对33 种目标物平均回收率的影响见图3。
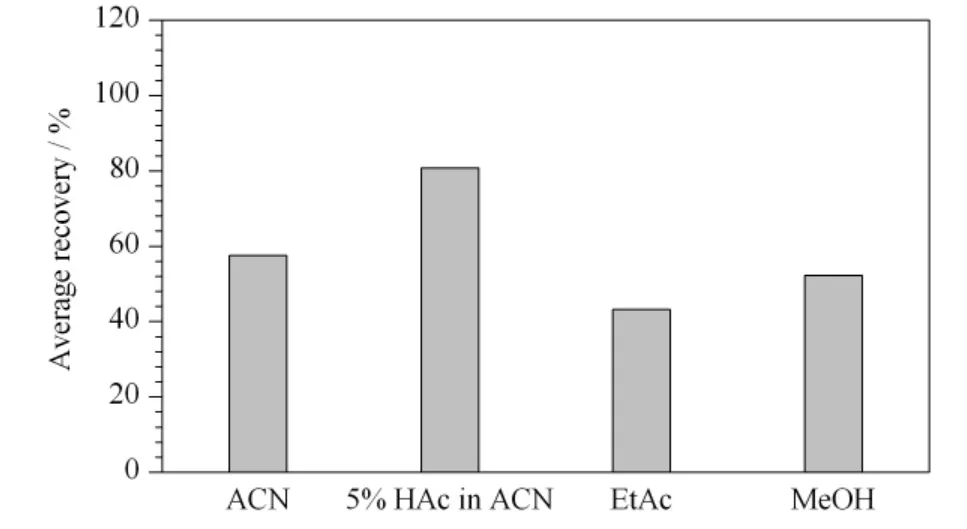
图3 不同提取剂对33 种目标物平均回收率的影响Fig.3 Effect of different extraction solvents on the average recoveries of the 33 analytes
对乙酸乙腈的酸度进行了优化,比较了1%、3%、5%、7%、10%、15% (v/v)乙酸乙腈的提取效率。结果显示,采用5% 和7% 乙酸乙腈提取时,回收率没有明显区别,提取效率最高,最终确定5%(v/v)乙酸乙腈为本方法的提取剂。
对提取剂的体积进行了优化,比较了不同体积(8、12、16、20、24、28 mL)乙酸乙腈的提取效率。结果表明,体积低于20 mL 时,随着提取溶剂体积的增加,回收率增大,当体积为20 mL 时,目标化合物的平均回收率最高为85.7%;随着提取剂体积的继续增加,共萃取干扰物增多,回收率呈降低趋势,因此最终选择20 mL 为本方法的提取剂体积。
2.4.3 吸附剂的优化
比较了C18、PSA、PAX、NH2、PEP 吸附剂及两两组合对提取液的净化效果及对目标化合物回收率的影响。C18 主要吸附非极性物质,对脂肪等共提物吸附性强,而NH2对糖类共提物净化效果好。从回收率角度看,C18、NH2两种吸附剂单独使用时,目标化合物的平均回收率分别为80.6% 和70.3%;当将200 mg C18 与200 mg NH2两种吸附剂组合使用时目标物的平均回收率为89.3%。表明两者混合使用时净化效果好于两者单独使用时的净化效果。因此最终确定吸附剂为C18 和NH2。不同吸附剂对33 种目标物平均回收率的影响见图4。
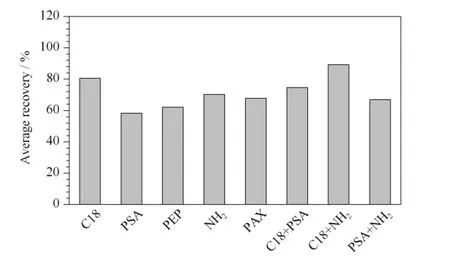
图4 不同吸附剂对33 种目标物平均回收率的影响Fig.4 Effect of different sorbents on the average recoveries of the 33 analytes
对吸附剂的用量进行了优化,考察了50 ~600 mg C18 与50 ~600 mg NH2按不同比例组合的净化效果及对目标化合物回收率的影响。结果表明,300 mg C18 与400 mg NH2组合时净化效果最好,回收率在70% ~120% 之间的目标化合物占比为90.9%,回收率在60% ~120% 之间的目标化合物占比为93.9%。因此最终确定C18 与NH2吸附剂的用量分别为300 mg 与400 mg。
2.5 方法考察
2.5.1 基质效应
通过比较空白基质匹配标准溶液和纯溶剂标准溶液响应值的差异评价方法的基质效应。参照文献[15],在空白基质提取液中添加混合标准溶液,与相同浓度纯溶剂混合标准溶液的信号强度(峰面积)相比,当比值等于或接近1 时(在0.85 ~1.15 范围内),认为基质效应不明显;小于0.85 时,表明存在明显的离子抑制作用,大于1.15 时表明存在明显离子增强作用。结果见图5,可见在猪肉样品基质中有63.6% 的目标化合物基质效应不明显,其余目标化合物都不同程度地存在离子抑制或离子增强基质效应。为尽可能地消除基质效应,实验采用基质匹配法进行定量分析。
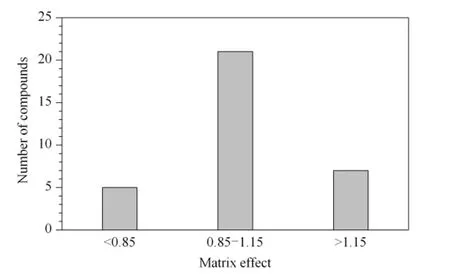
图5 猪肉样品基质中目标物的基质效应分布Fig.5 Distribution of matrix effect of the analytes in pork
2.5.2 方法的线性关系和定量限
取空白样品,按1.3 节方法进行提取、净化、定容得到样品提取液,根据目标化合物在质谱中响应的强弱,加入不同浓度的混合标准溶液。以准分子离子的峰面积Y 对含量X 做标准曲线,33 种目标化合物的线性范围及相关系数见表2。用空白基质提取液稀释标准溶液,直到获得每种目标化合物的信噪比等于10(S/N=10)的浓度,确定其为该化合物的定量限(LOQ)。
如表2 所示,33 种目标化合物在各自的线性范围内相关系数均大于0.99,LOQ 为2.5 ~100 μg/kg。对于大多数有最大残留限量(MRL)规定的兽药来讲,该方法的LOQ 小于MRL,可以满足快速筛查的需要。

表2 33 种目标化合物的线性关系和定量限Table 2 Linear relationships and limits of quantification of the 33 compounds
2.5.3 方法的回收率和精密度
在经测定不含有待测物质的猪肉样品中进行5个加标水平(LOQ、2LOQ、4LOQ、500 μg/kg 和1 000 μg/kg)的回收率和精密度测定,每个加标水平做6 个平行样,计算其回收率和相对标准偏差(RSD)。结果表明,33 种化合物的回收率在67.0%~109.0% 范围内,相对标准偏差均不高于15.1%(表略),表明方法回收率高、通用性强,可以满足日常猪肉样品中33 种兽药残留的同时测定。
2.6 方法应用
应用本方法对30 件从市场购买的猪肉样品进行检测,结果发现一件猪肉样品中检出土霉素,含量为90.5 μg/kg。为了验证该结果,我们采用国家标准方法GB/T 21317-2007《动物源性食品中四环素类兽药残留量检测方法 液相色谱-质谱/质谱法与高效液相色谱法》中的液相色谱-质谱/质谱法,对上述检出的阳性样品进行了确证,检测结果为土霉素97.0 μg/kg,与本文方法的测定结果相吻合。
3 结论
本文利用QuEChERS 方法提取、净化,结合高分辨的超高效液相色谱-四极杆-飞行时间质谱技术,建立了猪肉中6 类33 种兽药残留的检测方法。该方法快速、简便、灵敏,可以对样品中的目标化合物进行准确的定性和定量分析,适用于猪肉样品中多类兽药残留的快速检测。
参考文献:
[1] Xia M,Jia L,Ji Y P. Journal of Instrumental Analysis (夏敏,贾丽,季怡萍. 分析测试学报),2004,23(Z1):217
[2] Wang C Y,Wang Y P,Wang N,et al. Chinese Journal of Analytical Chemistry (王重洋,王远鹏,王宁,等. 分析化学),2013,41(1):83
[3] Bu M N,Shi Z H,Kang J,et al. Journal of Instrumental Analysis (卜明楠,石志红,康健,等. 分析测试学报),2012,31(5):552
[4] Wang L,Li Y Q,Wang H B,et al. Chinese Journal of Analytical Chemistry (王炼,黎源倩,王海波,等. 分析化学),2011,39(2):203
[5] Yan L Z,Zhao Y B,Fu Y,et al. Chinese Journal of Analysis Laboratory (晏利芝,赵永彪,富玉,等. 分析试验室),2011,30(1):2011
[6] Li N,Zhang Y T,Liu L,et al. Chinese Journal of Chromatography (李娜,张玉婷,刘磊,等. 色谱),2014,32(12):1313
[7] Cao Y F,Kang J,Chang Q Y,et al. Chinese Journal of Chromatography (曹亚飞,康健,常巧英,等. 色谱),2015,33(2):132
[8] Li X J,Peng T,Li C J. Journal of Instrumental Analysis (李晓娟,彭涛,李重九. 分析测试学报),2012,31(5):628
[9] Liu C,Chen Y,Li X W,et al. Chinese Journal of Food Hygiene (刘畅,陈燕,李晓雯,等. 中国食品卫生杂志),2014,26(5):464
[10] Peng L,Wu N P,Zhang C W,et al. Chinese Journal of Veterinary Medicine (彭丽,吴宁鹏,张崇威,等. 中国兽药杂志),2014,48(9):45
[11] Gao F D,Zhao Y,Shao B,et al. Chinese Journal of Chromatography (高馥碟,赵妍,邵兵,等. 色谱),2012,30(6):560
[12] Anastassiades M,Lehotay S J. J AOAC Int,2003,86(2):412
[13] Cao H,Chen X Z,Zhu Y,et al. Food Science and Technology (曹慧,陈小珍,朱岩,等. 食品科技),2013,38(6):323
[14] Wang W,Shi Z H,Kang J,et al. Chinese Journal of Analysis Laboratory (王伟,石志红,康健,等. 分析试验室),2013,32(4):82
[15] Wang L Q,He L M,Zeng Z L,et al. Journal of Chinese Mass Spectrometry Society (王立琦,贺利民,曾振灵,等.质谱学报),2011,32(6):321
