木糖醇和麦芽糖醇型亲水作用色谱固定相的制备及其分离性能的评价
2015-12-26肖红斌万伯顺
用 天, 吴 凡, 肖红斌,2* , 万伯顺*
(1. 中国科学院大连化学物理研究所,辽宁 大连116023;2. 北京中医药大学,北京100029)
亲水作用色谱(HILIC)对强极性和亲水性化合物具有很强的保留作用,作为反相液相色谱的一个重要补充,近些年受到越来越多的重视,业已广泛应用于各种极性和亲水性化合物的分离,如多肽和蛋白质[1,2]、糖类[2-5]、药物[6-8]、代谢物[9,10]、抗体[11]、毒物[12]、天然产物[13]和环境污染物[14]等的分离。
固定相是色谱的核心技术之一,亲水作用色谱固定相多以硅胶键合固定相为主,键合官能团主要包括氨基、二醇基、氰基、酰胺型、环糊精型、糖型、两性离子型和聚合物型等[15,16],其中醇羟基具有不电离、稳定性好和亲水性强等优点,是亲水作用色谱固定相最优秀的键合基团之一。早期的醇羟基类亲水作用色谱固定相是二醇基(diol)键合固定相,但是因其结构中的醇羟基比例较小而表现出较弱的亲水性和较强的疏水性[17-21],在亲水作用色谱领域的应用相对有限。与二醇基键合固定相类似,聚乙二醇(polyethylene glycol,PEG)键合固定相也表现出很强的疏水作用,其更多地应用于混合(亲水和反相)作用模式[17,22,23]。
为了克服早期醇羟基型亲水作用色谱固定相亲水性不足的缺点,近些年来相继有多种多羟基型亲水作用色谱固定相被开发出来,这些新型固定相因键合基团中含有较多的醇羟基而表现出很强的亲水性。2007 年,Liang 等[24-26]通过点击反应制备了葡萄糖(glucose)、麦芽糖(maltose)和β-环糊精(βcyclodextrin)修饰固定相,并成功将其应用于糖的分离分析;其中麦芽糖键合固定相展现出了十分出众的糖肽富集效应[27]。2008 年,Irgum 等[28]通过表面引发聚合的方式制备了亲水性更好的山梨糖醇(sorbitol)修饰固定相。2009 年,Huang 等[29]制备了壳寡糖(chitooligosaccharide)型固定相,并成功用于糖和糖肽的分离富集。2010 年,Marra 等[30]合成了半乳糖(ethynyl C-galactoside)和乳糖(propargyl O-lactoside)型亲水作用固定相,成功分离了单糖和二糖差向异构体。2011 年,Armstrong等[31,32]成功发展了基于环聚果糖6(cyclofructan 6)和硫酸化环聚果糖6 的新型亲水作用色谱固定相。2011 年,Lindner 等[33]通过美拉德反应(Maillard reaction)将还原性糖键合于氨基硅胶表面而成功制备了纤维二糖(cellobiose)型固定相。
糖醇是一大类多羟基型化合物,作为糖的衍生物,具有更好的化学稳定性和亲水性,十分有潜力作为亲水作用色谱固定相的键合基团,但目前除山梨糖醇已被应用于亲水作用色谱固定相外,鲜有文献报道其他种类的糖醇型亲水作用色谱固定相。为此,本文将线形的木糖醇(xylitol)和非线形的麦芽糖醇(maltitol)键合于硅胶表面,合成了两种新型的多羟基类糖醇亲水作用色谱固定相,并比较了两种固定相在分离性能和选择性上的异同。
1 实验部分
1.1 仪器与试剂
Agilent 1100 高效液相色谱仪(配紫外/可见光检测器、自动进样器,美国Agilent 公司);球形硅胶(粒径5 μm,比孔容0.74 mL/g,比表面积324 m2/g,日本富士公司;使用前先用10% (质量分数)的盐酸活化6 h,并于140 ℃下真空干燥8 h 后备用);乙腈(色谱纯,美国Sigma-Aldrich 公司);去离子水(电阻率>18 MΩ·cm,美国Milli-Q 水处理系统制备);异氰酸丙基三乙氧基硅烷(3-(triethoxysilyl)propylisocyanate,纯度95%)、乙酸铵(色谱纯)、乙酸(色谱纯)、木糖醇(纯度99%)、麦芽糖醇(纯度95%)(上海阿拉丁试剂公司);淫羊藿苷及其类似物(由本实验室制备并提供);吡啶(使用前用氢化钙干燥处理);其他试剂均为分析纯并直接使用。
1.2 糖醇键合硅胶的制备
合成路线如图1 所示。取5.0 g 干燥的木糖醇/麦芽糖醇溶于100 mL 无水吡啶中,加热至80℃,由滴液漏斗滴加6.8/3.0 g 异氰酸丙基三乙氧基硅烷后继续反应6 h;待反应液冷却后加入6 g 干燥硅胶,回流反应24 h;冷却后抽滤,依次用适量的吡啶、乙醇、水、乙醇和丙酮洗涤,最后于60 ℃下真空干燥6 h 即得到糖醇键合硅胶。

图1 木糖醇和麦芽糖醇键合硅胶的合成路线Fig.1 Synthesis route of xylitol/maltitol modified silica
1.3 色谱柱的填充及色谱分离条件
采用匀浆法将所制备的固定相装入250 mm×4.6 mm 的不锈钢色谱柱管中。乙酸铵水溶液的pH 用乙酸调节,死时间用甲苯测得,作为样品的化合物质量浓度为0.1 ~1 g/L。
2 结果与讨论
2.1 元素分析和基本柱评价
元素分析给出木糖醇键合硅胶的碳、氢元素的质量分数分别为7.76%、1.41%,麦芽糖醇键合硅胶的碳、氢元素的质量分数分别为8.21%、1.51%。碳元素含量的增加有力地证明了通过图1 所示的简单合成方法可以将糖醇分子成功地键合于硅胶表面。利用公式S=1 000×C/N×Mc(其中S 为糖醇基团键合密度,单位为mmol/g;C 为碳元素的质量分数;N为键合基团中的碳原子数;对于木糖醇和麦芽糖醇键合基团来说,N 分别为9 和16;Mc为碳原子的摩尔质量)计算糖醇的键合密度分别为0.72 mmol/g和0.43 mmol/g,木糖醇的键合密度约为麦芽糖醇的1.7 倍,产生这种差异的原因是相对分子质量较大的麦芽糖醇受到较大的空间位阻影响,因而键合密度明显小于相对分子质量较小的木糖醇。
利用3 种极性化合物(胸苷、胞嘧啶和鸟嘌呤)评价糖醇柱的保留能力、峰对称性和柱效。它们在木糖醇柱上的保留因子(k)分别为1.82、13.85 和17.51,峰不对称因子分别为0.79、0.91 和0.90,柱效约为16 000 N/m;在麦芽糖醇柱上的保留因子分别为2.06、11.22 和12.46,峰不对称因子分别为0.65、0.67 和0.69,柱效在21 000 ~24 000 N/m 之间。上述结果表明这两种糖醇柱对极性化合物都有很强的保留能力;相较之下,麦芽糖醇柱的柱效稍优于木糖醇柱,但峰对称性较木糖醇柱差,这是因为麦芽糖醇键合密度较低导致硅胶基质表面残留了较多的硅羟基。此外,在长时间使用中这两种糖醇柱均表现出一定的稳定性和重复性,如连续10 次进样分析,两种糖醇柱的保留时间的相对标准偏差均不超过0.3%。
2.2 流动相中乙腈含量对化合物色谱保留的影响
选择10 种极性和亲水性化合物作为测试样品,通过增加流动相中乙腈的含量(5% (v/v,下同)~95%)研究糖醇柱的保留机理。如图2 所示,在两种糖醇柱上,乙腈含量对保留因子的影响皆呈J 形曲线,这是典型的亲水作用色谱的保留曲线,一般认为其保留机理为分配机理。曲线拐点在乙腈含量为40% 附近,当乙腈含量小于40% 时,保留较弱且随乙腈含量的降低并未出现明显增加,而在亲水作用色谱主导区域(乙腈含量大于40%),保留会随乙腈含量的增加而显著增强。与其他化合物不同的是,对氨基水杨酸和淫羊藿苷在麦芽糖醇柱上仅以亲水作用为主(J 形曲线),但在木糖醇柱上表现为U 型曲线的特征,即亲水作用色谱和反相色谱混合模式,这是因为木糖醇键合硅胶因OH/CH(CH2)基团的摩尔比和极性基团数目均小于麦芽糖醇键合硅胶而表现出了一定的疏水(反相色谱)作用。

图2 流动相中乙腈含量对极性和亲水性化合物在(a)木糖醇和(b)麦芽糖醇柱上的保留影响Fig.2 Effect of acetonitrile content in the mobile phase for the retention of polar and hydrophilic analytes on (a)xylitol and (b)maltitol columns
2.3 模型混合物的分离和选择性比较
利用4 种极性/亲水性模型混合物评估木糖醇柱和麦芽糖醇柱的分离性能,模型混合物包含酸性、碱性和中性化合物以及电解质和非电解质,其中碱基及其相应的核苷是评估HILIC 柱最常用的模型化合物,利于与文献中报道的其他HILIC 固定相进行比较。图3 ~6 为4 种模型混合物在优化条件下的色谱图。总体来看,木糖醇柱能够很好地分离水溶性维生素、水杨酸及其类似物、碱基及其相应的核苷,而淫羊藿苷类似物、胸腺嘧啶和尿嘧啶在木糖醇柱上不能达到基线分离;相比之下,麦芽糖醇柱对这4 种模型混合物均有良好的分离效果,仅仅腺苷和腺嘌呤在麦芽糖醇柱上不能得到基线分离。
对于大多数测试化合物,木糖醇柱和麦芽糖醇柱表现出大致相同的选择性,只有维生素C 在木糖醇柱上先于维生素B3 洗脱,而在麦芽糖醇柱上后于维生素B3 洗脱(见图3)。同时麦芽糖醇柱表现出对水杨苷独特的选择性,在相同的色谱条件下,水杨苷在木糖醇柱上的洗脱顺序位于水杨酸和对氨基水杨酸之间,在麦芽糖醇柱上却在对氨基水杨酸之后,甚至落后于乙酰水杨酸(见图4)。考虑到非线形的麦芽糖醇分子比线形的木糖醇分子在化学结构上多一个环状的糖基,同时维生素C 和水杨苷在化学结构上也恰恰比同组化合物多一个环状(类)糖基结构,据此推测上述两点选择性的差异很可能来源于麦芽糖醇上的糖基和分析物的糖基之间强烈的氢键作用。这种推测可以进一步地被碱基及其相应的核苷的分离结果证明:核苷相对于其相应的碱基在分子结构上仅多一个环状糖基,以直链形的木糖醇柱为标准,选择因子αmaltitol/αxylitol的计算值(见表1)均大于1。类似地,表1 中淫羊藿苷及其类似物的αmaltitol/αxylitol计算值均大于1,也同样证明麦芽糖醇柱对环状糖基独特的选择性。此外,碱基及其相应的核苷在木糖醇和麦芽糖醇固定相上的洗脱顺序明显不同于其他多羟基型固定相[24,25,29,31,32],说明其具有独特的选择性。

图3 水溶性维生素在(a,b)木糖醇和(c)麦芽糖醇柱上的分离Fig.3 Separation of water soluble vitamins on(a,b)xylitol and (c)maltitol columns
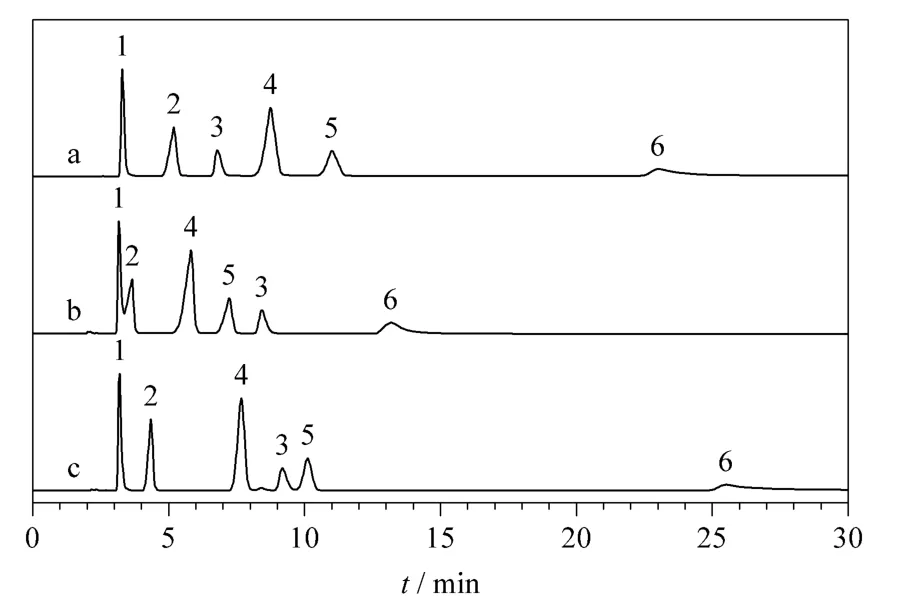
图4 水杨酸及其类似物在(a)木糖醇和(b,c)麦芽糖醇柱上的分离Fig.4 Separation of salicylic acid and its analogues on (a)xylitol and (b,c)maltitol columns
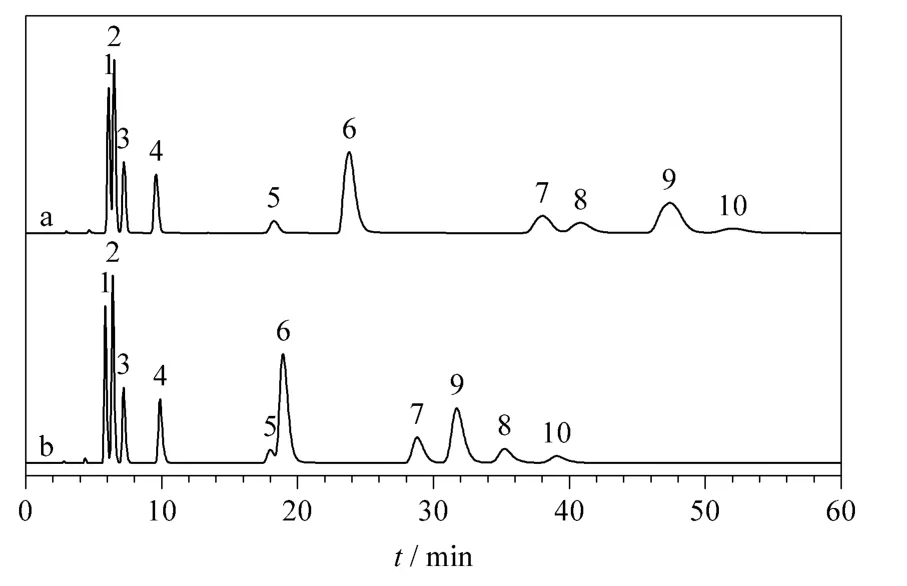
图5 碱基及其相应核苷在(a)木糖醇和(b)麦芽糖醇柱上的分离Fig.5 Separation of nucleic acid bases and nucleosides on (a)xylitol and (b)maltitol columns

图6 淫羊藿及其类似物在(a)木糖醇和(b)麦芽糖醇柱上的分离Fig.6 Separation of icariin and its analogues on(a)xylitol and (b)maltitol columns

表1 一些化合物及其糖基衍生物在两种糖醇柱上的选择因子比较Table 1 Comparison of selectivities of the analytes and its glycosyl derivatives on xylitol and maltitol columns
2.4 流动相pH 的影响
通过改变流动相pH(4.2 ~6.8)考察其对糖醇柱保留的影响。木糖醇和麦芽糖醇键合基团均为中性基团,一般不会受流动相pH 变化的影响,流动相pH 主要影响固定相表面的残余硅羟基(pKa≈4)和化合物的带电状态。一方面硅羟基会在较高的pH下逐渐解离而带有更多的负电荷,进而对带电的化合物产生越来越强的静电作用,另一方面化合物本身带电状态的改变会引起其亲水性的变化。如图7所示,在考察的pH 范围内,流动相pH 对木糖醇和麦芽糖醇柱的影响是相同的。当pH 从4.2 增大至6.8 时,硅羟基和维生素C(pKa=4.0)之间的静电斥力因电离程度的增加而增大,趋向于保留减弱,同时维生素C 因进一步电离而导致亲水性增强,趋向于保留增加,由于前者的影响大于后者,总的结果是维生素C 的保留减弱。pH 对维生素B3 和朝藿定B 的影响与维生素C 类似。相反的,维生素B6(pKa=5.0)的保留会随着pH 的增大而略有增加,这是因为一方面硅羟基的进一步电离有助于增加静电引力,另一方面维生素B6 的电离会趋向于静电引力和亲水性的减弱,从实验结果来看前一方面的原因略占优势。pH 对腺苷的影响与维生素B6 类似。对于水杨酸类化合物,pH 的增大会促进其电离,一方面增强了与硅羟基的静电斥力,另一方面增加了化合物的亲水性,对于水杨酸和对氨基水杨酸,前者的影响稍强于后者,故其保留略有减弱;而对于3,4-二羟基苯乙酸,前者的影响远不及后者,因而保留显著增加。另外,通过比较不同pH 下淫羊藿类似物的αmaltitol/αxylitol值(见表2),发现随着流动相pH 的增加,麦芽糖醇柱对糖基的选择性会有一定程度的增强。此外,流动相pH 的变化可以有效地改善糖醇柱的分离效果,如图3a 和3b 所示,维生素B2 和B6 的分离度随pH 的减小而稍有增加。
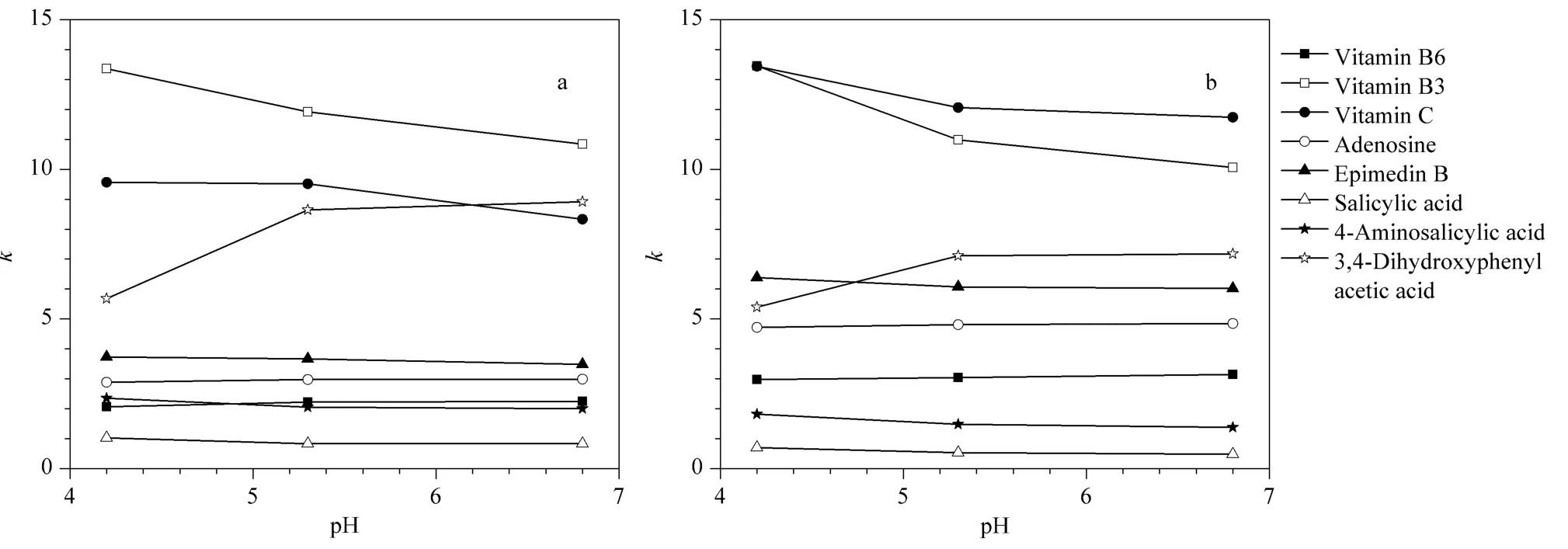
图7 (a)木糖醇和(b)麦芽糖醇柱上流动相pH 对化合物色谱保留的影响Fig.7 Effect of mobile phase pH for the retention factors of the analytes on (a)xylitol and (b)maltitol columns

表2 流动相pH 对糖基在麦芽糖醇柱上保留的影响Table 2 Effect of mobile phase pH for the retention of glycosyl on the maltitol column
2.5 缓冲盐浓度的影响
通过增加流动相中乙酸铵的浓度(5 ~40 mmol/L)考察缓冲盐浓度对保留的影响。如图8所示,在两种糖醇柱上缓冲盐浓度对化合物保留的影响是相同的,即化合物的保留均随着缓冲盐浓度的增加而增强。

图8 (a)木糖醇和(b)麦芽糖醇柱上缓冲盐浓度对化合物保留的影响Fig.8 Effect of buffer salt concentration for the retention of analytes on (a)xylitol and(b)maltitol columns
一般来说,流动相中缓冲盐浓度的增加一方面会引起吸附在固定相表面的水化层变厚而趋向于保留增强,另一方面也会引起更多的铵离子吸附在硅羟基表面,进而降低了硅羟基的平均带电量,导致其对分析物的静电斥力(或引力)减弱,趋向于保留增强(或减弱)。在糖醇柱上,对于中性的水杨苷来说,保留增强源于水化层的变厚;对于带负电的化合物如朝藿定B、对氨基水杨酸和维生素C 来说,保留增强是水化层增厚和静电斥力减弱共同作用的结果;对于带正电的化合物如维生素B2 和尿嘧啶来说,水化层增厚的影响大于静电引力减小的影响,总的作用结果仍是保留增强。另外,通过比较不同乙酸铵浓度下淫羊藿类似物的αmaltitol/αxylitol值(见表3),发现随着缓冲盐浓度的增加,麦芽糖醇柱对糖基的选择性变化不明显。此外,利用流动相中缓冲盐浓度的变化同样可以很好地改善糖醇柱的分离效果,甚至改变其选择性,如图4b 和4c 所示,在麦芽糖醇柱上水杨酸类化合物的分离度均随着乙酸铵浓度的增大而显著增加,洗脱顺序也会发生部分改变。

表3 缓冲盐浓度对糖基在麦芽糖醇柱上保留的影响Table 3 Effect of buffer salt concentration for the retention of glycosyl on the maltitol column
3 结论
本文制备了两种糖醇类新型多羟基亲水作用色谱固定相,这两种固定相对酸、碱和中性化合物以及电解质和非电解质,均表现出了良好的分离性能和较为新颖的选择性,特别是麦芽糖醇键合固定相显示出对糖基的独特选择性。这两种固定相为分离分析极性和亲水性化合物提供了新的可供选择的色谱固定相,具有很好的应用前景。
[1] Haberhauer-Troyer C,Delic M,Gasser B,et al. Anal Bioanal Chem,2013,405:2031
[2] Shen A,Li X,Dong X,et al. J Chromatogr A,2013,1314:63
[3] Fu Q,Liang T,Li Z,et al. Carbohydr Res,2013,379:13
[4] Liang T,Fu Q,Xin H X,et al. Chinese Journal of Chromatography (梁图,傅青,辛华夏,等. 色谱),2014,32(12):1306
[5] Fu Q,Wang J,Liang T,et al. Chinese Journal of Chromatography (付青,王军,梁图,等. 色谱),2013,31(11):1051
[6] Nemoto T,Lee X,Kumazawa T,et al. J Pharm Biomed Anal,2014,88:71
[7] Gallinella B,Bucciarelli L,Zanitti L,et al. J Chromatogr A,2014,1339:210
[8] Sun J,Zhang F,Peng Y,et al. J Chromatogr B,2013,913/914:55
[9] Kok M G M,Swann J R,Wilson I D,et al. J Pharm Biomed Anal,2014,92:98
[10] Wang X Y,Gao P,Xu G W. Chinese Journal of Chromatography (王希越,高鹏,许国旺. 色谱),2014,32(10):1084
[11] Kahsay G,Song H,Schepdael A V,et al. J Chromatogr A,2014,87:142
[12] Zhuo L,Yin Y,Fu W,et al. Food Chem,2013,137:115
[13] Guo X,Zhang X,Guo Z,et al. J Chromatogr A,2014,1325:121
[14] Qian F Z,Zhu L B,Xu N B,et al. Chinese Journal of Chromatography (钱飞中,朱丽波,徐能斌,等. 色谱),2014,32(5):535
[15] Garcia-Gomez D,Rodriguez-Gonzalo E,Carabias-Martinez R. TrAC-Trends Anal Chem,2013,47:111
[16] Jandera P. Anal Chim Acta,2011,692:1
[17] Jandera P,Hajek T. J Sep Sci,2009,32:3603
[18] Liu X,Pohl C. J Chromatogr A,2008,1191:83
[19] Wang X,Li W,Rasmussen H T. J Chromatogr A,2005,1083:58
[20] Marclay F,Saugy M. J Chromatogr A,2010,1217:7528
[21] Wu J Y,Bicker W,Lindner W. J Sep Sci,2008,31:1492
[22] Jandera P,Hajek T,Skerikova V,et al. J Sep Sci,2010,33:841
[23] Jandera P,Vynuchalova K,Hajek T,et al. J Chemometr,2008,22:203
[24] Guo Z,Lei A,Zhang Y,et al. Chem Commun,2007:2491
[25] Fu Q,Guo Z,Liang T,et al. Anal Methods,2010,2:217
[26] Fu Q,Liang T,Zhang X,et al. Carbohydr Res,2010,345:2690
[27] Yu L,Li X,Guo Z,et al. Chem Eur J,2009,15:12618
[28] Persson J,Hemstrom P,Irgum K. J Sep Sci,2008,31:1504
[29] Huang H,Jin Y,Xue M,et al. Chem Commun,2009:6973
[30] Moni L,Ciogli A,D′Acquarica I,et al. Chem Eur J,2010,16:5712
[31] Padivitage N L T,Armstrong D W. J Sep Sci,2011,34:1636
[32] Qiu H,Loukotkova L,Sun P,et al. J Chromatogr A,2011,1218:270
[33] Schuster G,Lindner W. Anal Bioanal Chem,2011,400:2539
