一步法分离石蜡切片DNA的改进与鉴定
2015-12-26,,,*
, , , *
(1.湖南省人民医院检验科,湖南 长沙 410005;2.湖南省人民医院临床输血科;3.中南大学生命科学学院分子生物学研究中心)
·技术与方法·
一步法分离石蜡切片DNA的改进与鉴定
彭剑桥1,雷平2,杨秦3,朱敏3*
(1.湖南省人民医院检验科,湖南 长沙 410005;2.湖南省人民医院临床输血科;3.中南大学生命科学学院分子生物学研究中心)
目的改进一种以含蛋白酶K的裂解液作用于石蜡包埋处理的细胞、分离DNA粗提液用于PCR扩增的实验方案。方法石蜡包埋组织切片经常规脱蜡处理,用蛋白酶K裂解液洗脱细胞,55 ℃消化3 h后离心,吸取上清液;分光光度法检测DNA酶裂解液质量和浓度;以之为模板分别扩增人线粒体基因微卫星多态D310区、ATPase6基因区和核基因HBB、BRCA1等的DNA片段,琼脂糖凝胶电泳检测分析PCR扩增情况,并测序验证扩增产物。结果以石蜡切片的DNA裂解粗提液为模板,扩增目的DNA片段阳性率可达100%;获得序列分析验证。结论改进的蛋白酶K裂解液一步洗脱消化法操作简单、过程快捷、费用低廉,能快速有效地分离石蜡组织切片DNA,用于后续PCR扩增检测,具有较高效率。
DNA分离; 石蜡包埋组织切片; 线粒体DNA; 核基因
越来越多的研究表明,人类多种复杂疾病如各种肿瘤、糖尿病、高血压、AD、PD、心血管疾病等的发生发展均与多重遗传因素相关,不仅包括核基因变异,也包括线粒体DNA(mitochondrial DNA,mtDNA)变异[1],同时,这些关联在不同人群中可能存在差别。为确证基因变异与不同疾病的相关性及其人群差异性,必须在人群中开展大规模的遗传学关联分析;而如果能对医院病理科积存的大量石蜡包埋组织切片加以有效利用,将有助于规避相关工作中耗时长的标本收集阶段,从而缩短研究进程、提高研究效率。从石蜡包埋组织切片中分离DNA或RNA虽然已经建立了比较成熟的方法[2-6],但大多程序繁琐,且试剂盒纯化系统价格不菲,对大样本处理而言劳动量大、费用高昂。本文根据文献[7]介绍的方法,运用含蛋白酶K的细胞裂解(消化)液直接洗脱和消化,对石蜡包埋处理的组织切片(包括新近制备和空气曝露5年以上的陈旧切片),分离其中细胞DNA成分,并进一步进行其PCR扩增应用的分析。
1 材料与方法
1.1样本石蜡包埋病理组织切片取自湖南省人民医院和中南大学湘雅三医院病理科。
1.2试剂二甲苯、无水乙醇、Tris粉等购自上海国药集团;蛋白酶K、Taq DNA聚合酶、dNTP等购自鼎国公司;DNA裂解消化缓冲液含0.5 mg/mL蛋白酶K、0.05 mol/L Tris-HCl(pH 8.5)、1 mmol/L EDTA和0.5% Tween-20[1]。
扩增包含人mtDNA D310 区(包含poly C微卫星多态)、部分ATPase6基因编码区(包含mtDNA单倍型特异性遗传标志8701A/G SNP位点)及核内的人乳腺癌易感风险基因1(breast cancer 1,early onset,BRCA1)和人β-珠蛋白(hemoglobin beta,HBB)基因部分片段的引物序列及扩增产物大小见表1。引物由北京鼎国公司合成。
表1PCR扩增引物

基因名称引物序列/退火温度片段大小ntDNA微卫生多态D310区5′⁃ACAATGAATGTCTTGCACAGC⁃CACTT⁃3′(上游)109bp5′⁃GGCAGAGATGTGTTTAAGT⁃GCTG⁃3′(下游)/60℃mtDNAATPase65′⁃AAGATTAAGAGAACCAACAC⁃CTCT⁃3′(上游)415bp5′⁃GTGGCAATAAAAATGATTAAG⁃GA⁃3′(下游)/58℃BRCAl5′⁃CATCTGGTAAGTCAGCA⁃CAAGAG⁃3′(上游)105bp5′⁃ACATGTCTTTTCTTCCCTAG⁃TATGT⁃3′(下游)/64℃HBB5′⁃GTGCACCTGACTCCTGAGGA⁃GA⁃3′(上游)102bp5′⁃CCTTGATACCAACCTGCCCAG⁃3′(下游)/58℃
1.3 DNA提取在通风橱内将载玻片上的石蜡切片以二甲苯静置洗脱约10 min,无水乙醇浸泡洗涤5 min×3次;脱蜡后的组织片变得疏松,室温下水平放置至无水乙醇完全挥发,然后以约180 μL消化液分次冲洗,将组织片转入1.5 mL离心管内,55 ℃消化3 h后沸水浴10 min以灭活蛋白酶K;12 000 rpm离心10 min后吸取上清液转入一洁净新离心管内,小心避免吸入沉淀残渣及悬浮的石蜡碎片;上清液即为所获DNA酶解溶液,各取1 μL在NanoDrop仪上进行DNA浓度测定和质量检测;余样置-20 ℃保存备用。
1.4 PCR扩增反应在20 μL PCR混合物中进行,其中含模板DNA约50 ng、200 μmol/L 4种dNTP、上游和下游引物各5 pmol、KCl 50 mmol/L、Tris-HCl (pH 8.3,25℃) 10 mmol/L、MgCl21.5 mmol/L及0.2 U Taq DNA聚合酶。扩增参数为94 ℃变性30 s、相应温度退火30 s,72 ℃延伸30 s,循环40次,最后于72℃充分延伸10 min。
1.5 PCR产物琼脂糖凝胶电泳检测及其测序分析取5 μL扩增产物分别进行2.5 %琼脂糖凝胶电泳检测,EB染色后紫外灯下观察结果,照相保留。
剩余PCR产物送至铂尚(Biosune)生物技术公司进行DNA序列分析。
2 结 果
2.1 DNA粗提液的紫外分光光度法检测通常如果在A260 nm处有最大吸收峰值,A260/280值大于1.8而小于2.0,表明样品DNA具有正常质量。部分石蜡切片样本DNA提取液质量检测结果显示:与对照(从正常人外周血白细胞分离的基因组DNA)不同,绝大部分样品的A260/280值仅为1.0左右,同时A260/230值极低(图1);此外,在吸收峰曲线中A260 nm处未出现明显的锐峰。
进一步对上述各样品取约100 ng进行琼脂糖凝胶电泳,以从正常人外周血白细胞分离的基因组DNA为对照,可见较分子量标志物23 130 bp位置略高的DNA条带,但全部10例石蜡切片DNA样品在各自泳道上未呈现DNA区带(图2)。
图1 10例不同石蜡切片DNA粗提液的质量检测结果 “control”为1例外周血白细胞基因组DNA

图2 10例不同石蜡切片DNA粗提液的琼脂糖凝胶电泳 M:λDNA/HindⅢ分子量标志物(小片段已电泳脱离凝胶); 1:对照(外周血白细胞基因组DNA); 2~11:10例不同石蜡切片标本DNA分离物(含DNA 100 ng)
2.2 PCR扩增的琼脂糖凝胶电泳检测经与50 bp DNA梯级分子量标志物比较,所获mtDNA D310区、ATPase6、HBB和BRCA1基因的PCR扩增产物大小与预期吻合(图3,表1)。但4对引物的扩增检测情况不尽相同,与BRCA1(图3D)相比,ATPase6(图3B)和HBB(图3C)的阳性率明显更高,而mtDNA D310区阳性率最低;此外,在标志物50 bp位置处多呈现一致密区域,系引物二聚体。
增加模板用量至100 ng后,mtDNA D310区扩增可见每个样本均获目的产物。
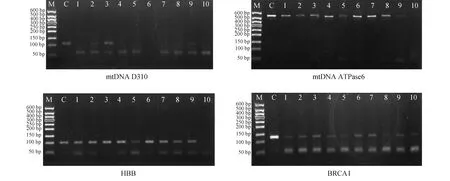
图3 PCR产物的琼脂糖凝胶电泳检测 M:50 bp DNA梯级分子量标志物;C:阳性对照;1~10:10个不同样本
2.3 PCR产物测序PCR产物直接测序结果进一步表明所获扩增物与设计相吻合。对部分具有明显识别标志(如D310 polyC微卫星多态区、mtDNA 8701A/G SNP位点)进行了搜索分析(图5)。
3 讨 论
根据文献[1]配制的DNA消化液中未使用强去垢剂SDS,而是较为温和的Tween 20,因为后者同样具有极强的破坏细胞膜功能但对后续PCR反应影响不大,同时,溶液中含有适当浓度蛋白酶K,从而能一步完成甲醛固定细胞的裂解及蛋白消化,促进切片上细胞DNA的分离。
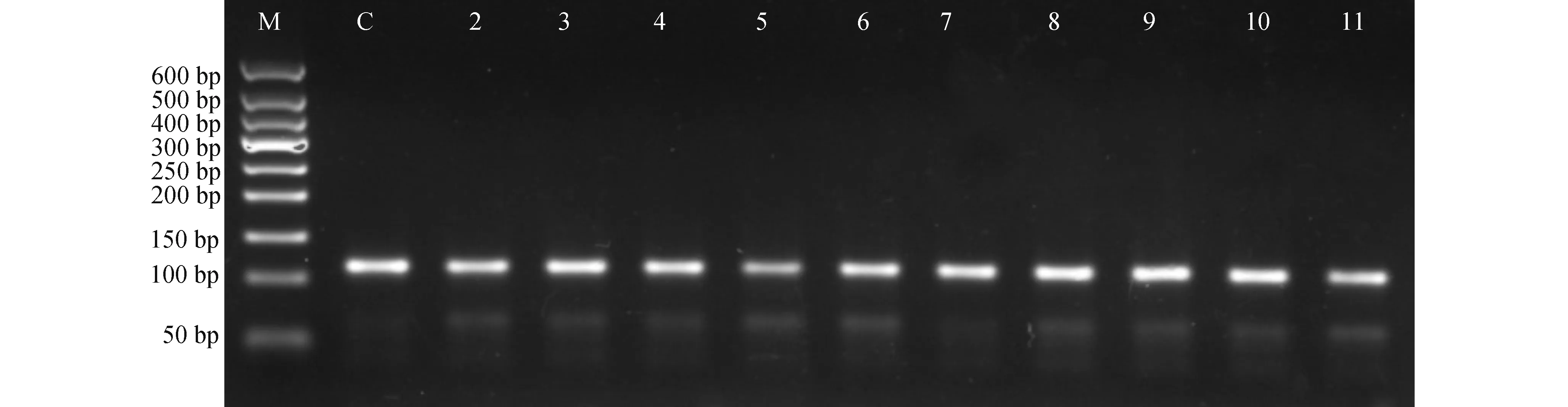
图4 模板DNA增量(至100 ng)后mtDNA D310区的PCR扩增检测 M:50 bp DNA梯级分子量标志物;C:阳性对照;泳道2~11:10个不同样本

图5 部分PCR产物的DNA序列分析结果 A:mtDNA D310区(下划线部分为poly C微卫星多态部分);B:mtDNA ATPase6基因(以下游引物测序结果,箭头所指处为8 701 G,系mtDNA宏单倍型M的标志SNP基因型之一)
DNA分离提取产率是制约石蜡包埋陈旧组织标本用于PCR扩增检测的难点之一。提取效率取决于两个方面:一是所用细胞数量,二是裂解消化的效率。最初在脱蜡处理后的玻片上直接滴加消化裂解液进行原位消化,结果在55℃较高温度、3 h较长时间处理过程中,因液体挥发严重,无法发生有效裂解和消化;因此改进为将脱蜡后组织片用消化裂解液洗脱并转移至离心管内,且每隔1小时低速瞬时离心富集液体,以补偿因水分挥发对细胞裂解和蛋白酶K消化作用的抑制。
针对切片上有限的细胞数量,为获得尽可能大的DNA产量,需用消化裂解液充分洗脱玻片上组织残片,尽量避免细胞的丢失。我们将180 μL溶液分三次冲洗收集细胞,每次用200 μL移液器吹出液体、冲击疏松的组织片,并使玻片一角伸入1.5 mL离心管管腔,使得液体汇聚后从角尖滴入管内。
由图2可以看出样本间浓度差别明显,这主要是因为切片上固定的组织细胞数量相互间还存在差别。DNA纯度根据A260/280比值判断,纯度高的DNA样品,其值在1.8左右,若比值高于1.9,说明DNA样品中RNA未除尽;若比值低于1.6,则表明样品中含有酚和/或蛋白质污染。本研究在组织消化后未进行纯化处理,因此DNA酶解分离液中含有大量蛋白质和有机类杂质,导致所有样本A260/280值均显著低于1.6,同时A260/230值均远小于2,提示高盐离子浓度;琼脂糖凝胶电泳中未出现DNA条带的原因在于切片DNA主体为片段化的核酸分子,大小不一,因此无法簇集形成致密区带。然后,由于后续PCR实验对模板DNA的纯度要求并不高,且所扩增多为小DNA片段(多为100 bp左右,不超过500 bp),因此适用此方案;若对DNA样品有纯度要求,可用常规方法如酚/氯仿法、柱层析法等进一步抽提纯化,但推测将造成DNA得率的下降。
由图3可以看到,在PCR扩增验证实验中,绝大部分样品获得4对引物相应的扩增产物,虽然阳性率和强度不一;而部分样品扩增产物带极弱或呈阴性的原因,推测有二:①切片上细胞数量过少,导致分离所获DNA产量极低下,如样品5,浓度仅为9.3 μg/mL,与其他样品相差达数倍至数十倍,故其4种PCR产物均相对较少,条带弱;②模板DNA降解,导致起始分子数过少、模板效用性过于低下所致。如样品9和10,尽管表观测定浓度分别达到75.9 μg/mL和47.4 μg/mL,但扩增条带弱甚至呈阴性。但从总体来看,通过增加模板用量可提高扩增阳性率、增加产物量,如图4所示,针对相同条件下mtDNA D310区扩增阳性率偏小的情况,增加1倍的模板用量至100 ng/20 μL反应,结果全部样品均获得理想的扩增检测结果。
引起石蜡包埋组织细胞DNA降解的原因来自多个方面。在组织块经历甲醛固定和高热石蜡冷却包裹的过程中,细胞内DNA易受损伤,加之所用切片多在空气中曝露达1年以上,这可能是造成DNA严重降解、所提取多为碎片化较小DNA分子并导致其用于PCR扩增时模板效率下降的主要原因。但是,这种影响对细胞质中线粒体DNA和核基因有较大差别:每个细胞具有数十至数百个线粒体,每个线粒体可拥有1个以上mtDNA分子,因而细胞内mtDNA可达数百到上千个拷贝,同时分子尺寸较小—人类线粒体DNA为仅16 569 bp的环状分子;而核基因多系细胞内单个拷贝,且为长链大分子,加之二甲苯法虽脱蜡较为彻底,但容易导致DNA长链断裂、降解;因DNA拷贝数量和分子大小的双重因素,mtDNA在耐受降解上具有相对核基因的显著优势。当然,PCR扩增效率并非完全取决于模板效用性,还与引物及待扩增区域的结构等有关,本文中D310区的扩增在其他条件一致的情况下反而较核基因如HBB更差可能属于后者。
本研究结果表明,改进的蛋白酶K裂解液“一步法”能快速从石蜡包埋组织切片中分离获得细胞DNA,因不经纯化回收处理,不仅产率不致严重受损,且规避了繁琐的纯化处理过程和昂贵的试剂费用;即使来自陈旧标本DNA粗提液也可有效用于PCR扩增线粒体基因片段。因此,医院病理科历年来所积存的病理组织包埋块或切片都可提供作为至少线粒体DNA疾病遗传学关联研究的重要标本来源,同时,mtDNA序列表征人类母系遗传背景,对于个体鉴别等也具有重要意义,而犯罪现场或灾难事故现场来源的陈旧血迹、毛发等核酸包含材料,完全有可能视为石蜡切片的情况,可进行相似的高效、快速核酸分离。
综上,尽管石蜡切片上经高温和有机溶剂处理获取DNA数量有限、且质量难以控制,但用于某些定性研究工作如基于PCR短片段扩增的个体基因分型,尤其对线粒体DNA的相关工作,仍然提供了来源丰富而合理的优质资源。
[1] Zhou WM,Zhu M,Gui M,et al.Peripheral blood mitochondrial DNA copy number is associated with prostate cancer risk and tumor burden[J].PLoS One,2014 9(10):e109470.doi:10.1371/journal.pone.0109470.eCollection 2014.
[2] Yuan YT,Jiang YX,Yin XW,et al.Comparison of four methods for DNA extraction from paraffin-embedded tissues[J].J Clinic Rehabilitative Tissue Engineering Res,2010,14(24):4430-4434
[3] 张素娟,赵彤,董敬朋,等.石蜡切片提取方法的改良及其临床应用临床[J].临床与实验病理学杂志,1996,R446:62-65.
[4] 赵吉生,盖宝东,森隆弘,等.石蜡包埋组织中提取DNA应用于PCR 反应影响因素的研究[J].中国实验室诊断杂志,2003,7:330-333.
[5] 宋丽新,高兴华,张士发,等.石蜡包埋组织中DNA提取方法的对比研究[J].中国麻风皮肤病杂志,2006,22(4):268-269.
[6] 白春英,郝俊海,白海花.石蜡包埋组织中DNA的提取[J].生物学杂志,2002,19(3):35-36.
[7] Aral C,Kaya H,Ataizi-Celikel C,et al.A novel approach for rapid screening of mitochondrial D310 polymorphism[J].BMC Cancer,2006,6:21-25.
DNAIsolationfromParaffinSectionsbyanAdjustedOne-StepProtocolanditsQualificationTestbyPCR
PENG Jianqiao,LEI Ping,YANG Qin,et al
(ClinicalLaboratory,HunanProvincialPeople’sHospital,Changsha,Hunan410005,China)
ObjectiveTo improve a simple method to extract DNA from tissues fixed in paraffin sections by direct digestion with proteinase K lysis buffer.MethodsFormalin-fixed paraffin-embedded sections were routinely deparaffinized and then the tissues were washed off for the glass slides using digestion buffer with proteinase K and transferred into a 1.5 mL microtube for each sample.After incubation at 55 ℃ for 3 h,the supernatant containing DNA was transferred into a new tube.The DNA concentration and A260/280values of the lysate were assayed by ultraviolet spectrophotometry.Amplification of the mitochondrial DNA fragment flanking D310 microsatellite region,part of the ATPase6 gene as well as nuclear genes HBB and BRCA1 were performed,and PCR amplification following detection with agarose gel electrophoresis was analyzed.ResultsAs shown by agarose gel electrophoresis and DNA sequence analysis,all the target mtDNA fragments were successfully amplified by PCR using the DNA lysate directly as template.ConclusionA simple-manipulation,rapid-procedure and low-cost method for DNA separation from paraffin sections was presented here,by which the DNA content was extracted by one-step digestion with proteinase K lysis buffer;its effectiveness were evidenced by PCR of mitochondrial DNA and nuclear DNA fragments.
DNA extraction; paraffin-embedded sections; mitochondrial DNA; nuclear gene
10.15972/j.cnki.43-1509/r.2015.06.027
2015-08-21;
2015-10-22
湖南省科技计划(2010FJ3084);中南大学学位与研究生教育教学改革研究(2340-74333003019);中南大学教师科研基金(2013JSJJ042).
*通讯作者,E-mail:zhumin0678@yahoo.com.
R446
A
(此文编辑:朱雯霞)
