固相萃取–气相色谱法测定甘草中的六六六、滴滴涕含量
2015-12-24张海燕
张海燕
(新疆库尔勒出入境检验检疫局,新疆库尔勒 841000)
甘草,气微,味甜而特殊[1],是一味传统中药材,也是新疆重要的进出口商品,主要用于药品、食品、化妆品及香烟类行业。六六六、滴滴涕(DDT)等有机氯农药,化学性质稳定,脂溶性大,残效期长,易在脂肪组织中蓄积,造成人体慢性中毒,严重危及身体健康[2]。尽管我国已在1983 年禁止使用六六六、滴滴涕等有机氯农药,但这些农药至今在土壤、地下水等环境中仍有残存[3]。随着中草药国际贸易的日益频繁,甘草质量越来越受到重视,建立甘草中六六六、滴滴涕农药残留量的快速检测方法十分必要。
目前关于测定上述两种农药残留量的报道较多,但对甘草中六六六、滴滴涕农药残留检测进行研究的报道不多[4–6]。此类农药的前处理方法主要有硫酸磺化法[7]、全自动凝胶净化法(GPC)[8–9]、Quchers法[10]、固相微萃取法[11]等。以上方法检测周期长,操作步骤相对繁琐,试剂消耗量较大,提取效率不高,难以满足大批量、多批次检测的需求[12]。
笔者采用固相萃取法以Florisil 柱净化,气相色谱–电子捕获检测器经DB–1 毛细管柱分析,建立了甘草中农药六六六、滴滴涕残留量的测定方法,该方法简便、省时、准确可靠,完成全部分析仅需15 min。
1 实验部分
1.1 主要仪器与试剂
气相色谱仪:7890A型,配63Ni电子捕获器(ECD),美国Agilent 公司;
涡旋混合器:ms3型,德国IKA公司;
超声波清洗器:SB5200型,必能信超生(上海)有限公司;
腕式振荡器:WX 51100–35型,美国Burrell公司;
氮吹仪:N–EVAP 112型,美国Organomation Associates公司;
离心机:CF16RXII型,日本HITACHI公司;
固相萃取装置:美国Waters公司;
旋风磨:CYCLOTEC 1093型,丹麦Foss公司;
电子天平:XP205型,瑞士Mettler Toledo公司;
固相萃取小柱:弗罗里硅土柱(Florisil),1 000 mg/(6 mL),美国Agilent 公司;
α-六六六、β-六六六、γ-六六六、δ-六六六、P,P’-DDE(滴 滴 伊)、P,P’-DDD(滴 滴 滴)、O, P-DDT、P,P’-DDT标准物质:纯度分别不小于97.5%,97.5%,98.5%,99.6%,98.0%,99.5%,98.5%,99.5%,Dr. Ehrenstorfer 公司;
氯化钠:分析纯,天津风船化学试剂科技有限公司;
正己烷、乙腈、丙酮:色谱纯,美国Burdick & Jackson公司;
实验用水为超纯水。
1.2 色谱条件
色谱柱:DB–1型弹性石英毛细管色谱柱(30 m×0.25 mm,0.25 μm);进样口温度:220℃;检测器温度:320℃;进样方式:脉冲不分流;载气:高纯氮(纯度不小于99.999%),流量为1.0 mL/min;尾吹气:氮气,流量为60 mL/min;进样体积:1 μL;程序升温:初始温度为70℃,保持1 min后,以20 ℃/min升至250℃,保持5 min,再以20℃/min升至270℃。
1.3 样品处理
1.3.1 提取
将甘草样品切断后置于旋风磨中粉碎,准确称取1.00 g甘草粉碎制品,加入10 mL水浸泡30 min,再加入20 mL乙腈,于旋涡仪上涡旋混匀1 min,超声20 min,震荡20 min,加入2.00 g NaCl进行盐析,以10 000 r/min离心5 min,取上清液10 mL (相当于0.5 g样品),用N2吹干,加入2 mL正己烷,溶解,待净化。
1.3.2 净化
将Florisil小柱置于固相萃取装置上,小柱先用5 mL丙酮–正己烷(1∶9)活化,用正己烷5 mL平衡,然后加入上述2 mL待净化样品液,用8 mL丙酮–正己烷(1∶9)分4 次洗脱,每次2 mL,收集全部洗脱液于10 mL刻度试管中,用氮气吹干后加入1 mL正己烷溶解,定容,上机,以气相色谱法测定。以保留时间定性,色谱峰面积外标法定量。
1.4 标准溶液配制
农药标准储备液:精密称取0.01 g(精确至0.1 mg)标准物质于10 mL容量瓶中,用丙酮–正己烷(1∶9)定容,配制成约1 000 μg/mL单标标准储备液,于–18 ℃保存。
混合农药标准溶液:各取适量体积的对应农药标准储备液于10 mL容量瓶中,用正己烷稀释定容,配制成10 μg/mL混合标准中间液。临用前用正己烷逐级稀释配制成对应浓度的混合农药标准溶液。
2 结果与讨论
2.1 提取溶剂的选择
常用的提取溶剂有乙腈、丙酮和乙酸乙酯。丙酮提取过程中会将大量色素同时提取出来,影响后续净化;乙酸乙酯与水不混溶,不易渗入植物细胞,不利于农药萃取[13];乙腈作提取溶剂,渗透性好,在盐饱和状态下可以与水完全分离,应用于气相色谱分析有很好的适用性。故选择乙腈作为提取溶剂。
考察加水与不加水条件下农药的回收率,试验结果表明加水浸润提取的回收率高于不加水提取的回收率,这可能是因为浸润后水分子能减少样品颗粒之间的吸附力,可使提取溶剂更好地浸入样品,有利于样品内农药的释放,从而增大提取效率。因此在用有机溶剂提取前向甘草样品中加适量水浸润。
2.2 固相萃取柱的选择
N-丙基乙二胺(PSA)主要去除糖类、脂肪酸、有机酸、酚类和亲脂性色素等极性物质;C18柱是QuEChERS 方 法[14](Quick,Easy,Cheap,Effective,Rugged,Safe)最为重要的净化剂,除去脂肪、维生素、色素、甾醇效果较好。相对于弗罗里硅土(Florisil ),C18和PSA对农药的回收率影响很小,但它们对色素的净化效果均不理想。石墨化炭黑(GCB)主要去除色素等物质,但在多残留农药的检测中会导致回收率下降,因此在无颜色或者浅颜色的基质中使用较少。Florisil主要用于分离氯化杀虫剂,在美国分析化学家协会(AOAC)和美国环保署(EPA)等推荐方法中也多用它[15],基于上述原因和目标物的特性,选择弗罗里硅土固相萃取柱为净化柱。
2.3 洗脱溶剂
洗脱剂的选择关系到测定结果的准确度。若洗脱剂极性太强,会洗下一些强保留的杂质组分;若洗脱剂极性太弱,则有些待测组分难以被完全洗脱[16]。用丙酮–正己烷(1∶9)调节洗脱剂的极性效果较好,因此选择丙酮–正己烷(1∶9)为洗脱剂,洗脱剂的用量为8 mL,流量为2.0 mL/min。
2.4 标准曲线和检出限
将10 μg/mL混合农药标准中间液稀释成1.0 μg/mL的混合标准溶液,并以此分别配制质量浓度为0.005,0.01,0.02,0.05,0.10,0.20,0.50 μg/mL的系列混合标准工作溶液,进样分析。各组分浓度均为0.1 μg/mL混合标准溶液的色谱图见图1。以色谱峰面积(y)对农药质量浓度(x)进行回归,计算得相应农药的回归方程。以3倍信噪比时的添加浓度作为方法检出限,以被测组分的信号值大于基线平均噪音10倍时的浓度为定量限,线性范围、线性回归方程及相关系数见表1。
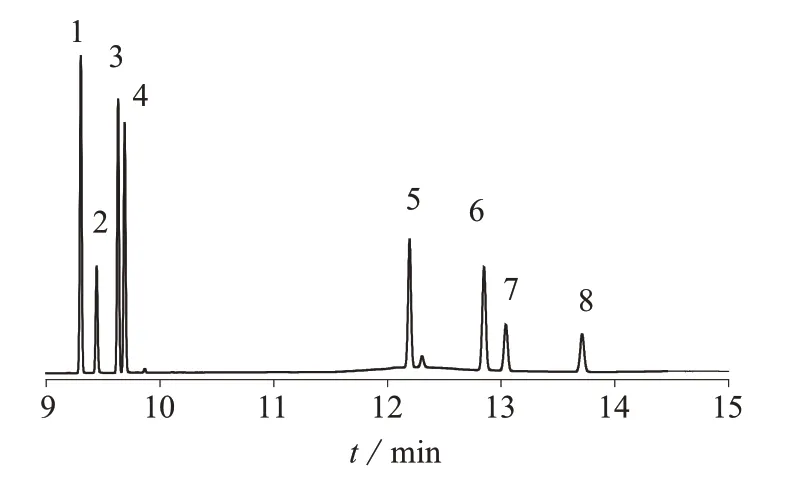
图1 混合农药标准溶液色谱图

表1 线性范围、线性回归方程、相关系数和检出限
2.5 方法精密度和回收率
称取空白甘草样品2.00 g,按标准加入法添加相当于含量0.05,0.1,0.5 mg/kg 3个水平的BHC,DDT 混合标准溶液,按1.3方法处理样品,在1.2色谱条件下测定,每个添加水平重复5 次,得2 种农药共计8 种同分异构体的回收率。空白样品及加标样品的色谱图分别见图2、图3。回收率和相对标准偏差结果见表2。
图2 空白样品色谱图

图3 加标样品(添加0.05 mg/kg)色谱图
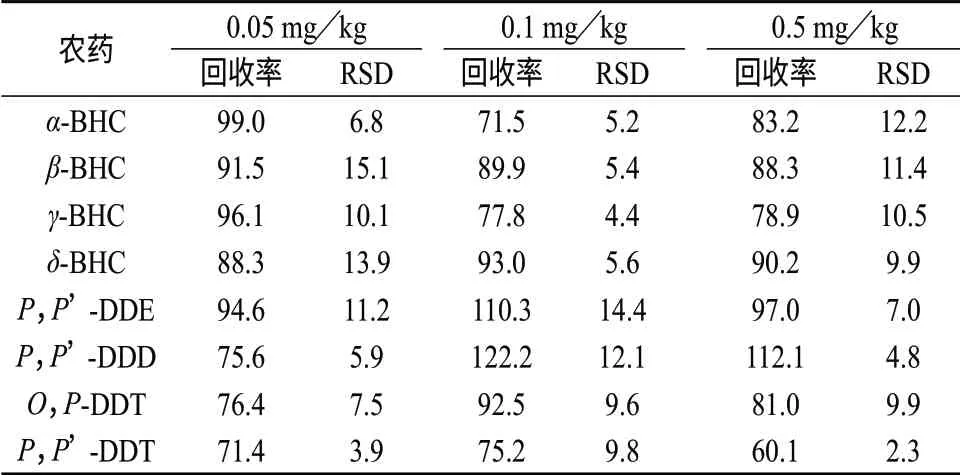
表2 空白甘草样品中六六六、滴滴涕农药的回收试验结果(n=5) %
由表2可知,在3个添加水平下,六六六、滴滴涕8个异构体,除P,P’-DDT 的回收率为60.1%外,其余回收率在71.4%~122.2%之间,相对标准偏差(RSD)为2.3%~15.1%,方法的精密度和准确度均符合残留分析的要求。
3 结语
建立了甘草中六六六、滴滴涕农药及异构体的多残留气相色谱检测方法。样品以乙腈为提取剂,水浴超声辅助震荡提取,弗罗里硅土固相萃取柱净化,DB–1 毛细管色谱柱分离,电子捕获检测器检测。该方法的灵敏度和准确度均满足实际样品检测的需要,方法简便,测定快速。
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2010: 80–81, 附录IX Q.
[2]宋家玉,李泽国,陈金东,等.填充柱气相色谱法测定食品中有机氯农药[J].化学分析计量,2007,16(3): 37–39.
[3]傅巧真,路俊仙,王萌,等.中药材农药残留原因及防治措施的研究进展[J].时珍国医国药,2014,25(4): 925–927.
[4]杨红兵,张玲,齐誉.新疆甘草中有机氯农药残留量检测研究[J].时珍国医国药,2006,17(11): 2 143–2 144.
[5]张曙明,郭怀忠,陈建民.甘草中有机氯类农药残留量的毛细管气相色谱测定[J].药学学报,2000,35(8): 596–600.
[6]王会丽,陈建民,张曙明,等.黄芪、甘草中有机氯农药残留量的气相色谱检测[J].药物分析杂志,1998,18 (5): 325–329.
[7]陈骁鹏,叶慧,仇雅静,等.泰州地区7种中药材中的有机氯农药残留量的测定[J].药物分析杂志,2012,32(7): 1 196–1 204.
[8]谢耀轩,王淑红,王铁杰,等.分散固相萃取–在线凝胶渗透色谱–气相色谱–质谱法检测香港中成药中20种有机氯农药残留[J].沈阳药科大学学报,2014,31(7): 535–541.
[9]何睿,刘雪峰,刘海静.中药材中20种农药残留的检测方法研究[J].药物分析杂志,2011,31(11): 2 164–2 167.
[10]吴剑威,徐荣,赵润怀,等.QuEChERS–气相色谱法快速检测五十种中药材中九种有机氯农药残留的方法研究[J].分析科学学报,2011,27(2): 167–170.
[11]董春洲,王文芳.顶空固相微萃取气相色谱法测定萝卜中有机氯农药及类似物[J].化学分析计量,2007,16(2): 16–19.
[12]刘桂宁,强维,钟闱桢,等.固相萃取/毛细管气相色谱法测定肉桂皮中六六六、滴滴涕农药残留量[J].分析测试学报,2014,33(1): 103–107.
[13]李鑫健,方翠芬,马临科,等.气相色谱-质谱联用法测定浙贝母中20种农药残留[J].药物分析杂志,2012,32(3): 419–423.
[14]胡西洲,程运斌,胡定金.农药多残留分析中QuEchERS 方法介绍[J].现代农药,2006,5(4): 24–44.
[15]程志,张蓉,刘韦华,等.气相色谱-串联质谱法快速筛查测定中药材中144种农药残留[J].色谱,2014,32(1): 57–68.
[16]班睿,郑建立,黄荣茂,等.固相萃取–毛细管气相色谱法测定中药材中有机氯农药残留[J].贵州农业科学,2006,34(5): 12–14.
