Acute coronary syndrome: a rare case of multiple endocrine neoplasia syndromes with pheochromocytoma and medullary thyroid carcinoma
2015-12-15AlessadroMalobertiPaoloMeaniRobertoPirolaMarisaVarrentiMarcoBoniardiAnnaMariaDeBiasePaolaVallerioEdgardoBonacinaGiuseppeManciaPaolaLoliCristinaGiannattasio
Alessadro Maloberti, Paolo Meani,, Roberto Pirola, Marisa Varrenti,, Marco Boniardi, Anna Maria De Biase,Paola Vallerio, Edgardo Bonacina, Giuseppe Mancia, Paola Loli, Cristina Giannattasio,
1Health Science Department, Milano-Bicocca University, Milan 20159, Italy; 2Cardiology IV, “A.De Gasperis” Department,Ospedale Niguarda Ca’ Granda, Milan 20159, Italy; 3General Oncologic and Mini-invasive Surgery Department, 4Anatomy and Histology Department, 5Endocrine Unit, Niguarda Ca’ Granda Hospital, Milan 20159, Italy
Introduction
Pheochromocytoma, a tumor arising from neuroectodermal chromaffin tissues, produces, stores, and secretes catecholamines1.The prevalence of the tumor is approximately 0.1%-0.6% in the hypertensive population2, of which the majority of the cases arise from adrenal medulla and 10%-20% from extraadrenal paraganglia (paragangliomas)3.Approximately 10%-20% of pheochromocytomas are malignant4, and genetic factors are implicated in 10%-20% of the cases, particularly in Von Hippel–Lindau, multiple endocrine neoplasia type 2, and neurofibromatosis5,6.
Typical presentations of pheochromocytoma include episodic headache, sweating, and tachycardia.Several patients with pheochromocytoma manifest paroxysmal hypertension and hypertensive crisis with sustained hypertension or normal blood pressure (BP)1.Other patients present cardiovascular events,including cardiac arrhythmias (sinus tachycardia or bradycardia and supraventricular arrhythmias), heart failure by toxic cardiomyopathy, angina, and acute coronary syndrome (ACS),in the absence of coronary artery diseases7.Myocardial ischemia is mediated by adrenergic mechanisms, including catecholamine induction of myocardial oxygen demand and coronary vasospasm.Patients with pheochromocytoma present a 14-fold higher risk of cardiovascular events than that of hypertensive cases8,9.
Although several cases and retrospective analyses described the diagnosis of pheochromocytoma after ACS presentation10-20,only a few studies reported this condition in the setting of hyperthyroidism and/or genetics21,22.
Case report
A 49-year-old Asian man with oppressive chest pain associated with hypertensive crisis (BP, 200/100 mmHg), with no other symptoms, was referred to our hospital.The patient’s medical history included hypertension and smoking.Sixteen years prior to the current condition, the patient had undergone surgical mitral and aortic valve substitution with mechanical prosthesis and followed by re-substitution because of secondary thrombosis.He had a history of paroxysmal atrial fibrillation, which was effectively controlled using 200 mg of amiodarone.However, the medication was stopped 2 weeks before the admission because of evidence of thyrotoxicosis [thyroid-stimulating hormone, 0.008 μIU/mL;free triiodothyronine (fT3), 8.6 pg/mL; and thyroxine (fT4),43.0 pg/mL], which was treated with steroid, potassium perchlorate, and methimazole.
Upon admission, the patient was oriented and alert, with a BP of 200/100 mmHg, heart rate of 100 bpm, and normal physical examination results.The electrocardiogram showed sinus tachycardia and non-specific ST-segment and T-wave changes.Laboratory examination results showed increased levels of myocardial necrosis enzymes (troponine T-hs, 133 ng/L; and creatine kinase−MB isoenzyme, 13.0 ng/L) and thyroid hormones(TSH, 0.007 μIU/mL; fT3, 8.3 pg/mL; and fT4, 44.2 pg/mL).
After the patient was initially diagnosed with ACS, echoscope was performed and showed a modest and diffuse decrease of left ventricular contraction with a conserved ejection fraction(0.50) but color Doppler analysis indicated normal heart valves.Coronary angiography did not show any clinically relevant coronary lesion, whereas thyroid echography demonstrated a heterogeneous structure in the absence of cysts and nodules.Without evidence of coronary diseases but with initial response to thyreostatic and steroid administration (fT3, 5.4 pg/mL; and fT4 36.2 pg/mL), the patient was finally diagnosed with ACS caused by hyperthyroidism.The patient was discharged and followed up by an endocrinologist and a cardiologist.
After 5 months, the patient was hospitalized again because of chest pain associated with hypertensive crisis (BP, 280/120 mmHg),nausea, headache, and vomiting.He was unresponsive to therapy with oral beta-blockers, alpha-blockers, and clonidine.
As the patient’s clinical case history was highly suggestive of pheochromocytoma, bioassays were performed.The following results were obtained: 24-h urinary epinephrine, 547 μg/24 h; 24-h urinary 633 μg/24 h; 24-h urinary metanephrine, 8,720 μg/24 h; 24-h urinary, 4,120 μg/24 h; serum calcitonin, 312 pg/mL; and normal parathormon, 34 pg/mL.
A subsequent whole-body computed tomography (CT)showed a 35 mm right adrenal lesion (Figure 1, Figure 1A)with smooth margins and a density higher than 2 HU.The diagnosis was right adrenal pheochromocytoma with suspected medullary thyroid carcinoma (MTC) with the MEN2A syndrome.After collegial discussion, the patient was treated with right adrenalectomy and thyroidectomy, as well as central compartment lymphadenectomy and parathyroid exploration.
Macroscopic examination indicated normal results.The thyroid sample showed intra-parenchymal isolation of the right lobe, with histological features suggestive of MTC(Figure 2).No lymphatic and vascular invasion was detected,and the surgical margins were free of tumor.The histological examination of the adrenal gland (Figure 1B) showed evidence of pheochromocytoma without histological malignant characteristics, specifically necrosis, atypical mitoses, and vascular invasion.Invasion of capsular and peri-adrenal tissues was not detected, and rare tumor cells exhibited intense positive cytoplasmic staining for calcitonin.
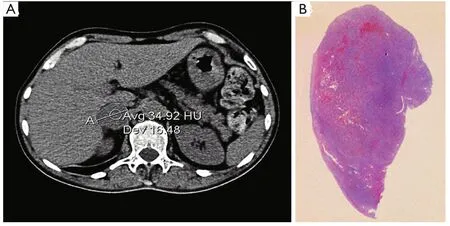
Figure 1 (A) Pheochromocytoma: abdominal computed tomography showed a 35 mm right adrenal lesion with smooth margins and a density higher than 2 HU.(B) Histological findings, H&E staining.
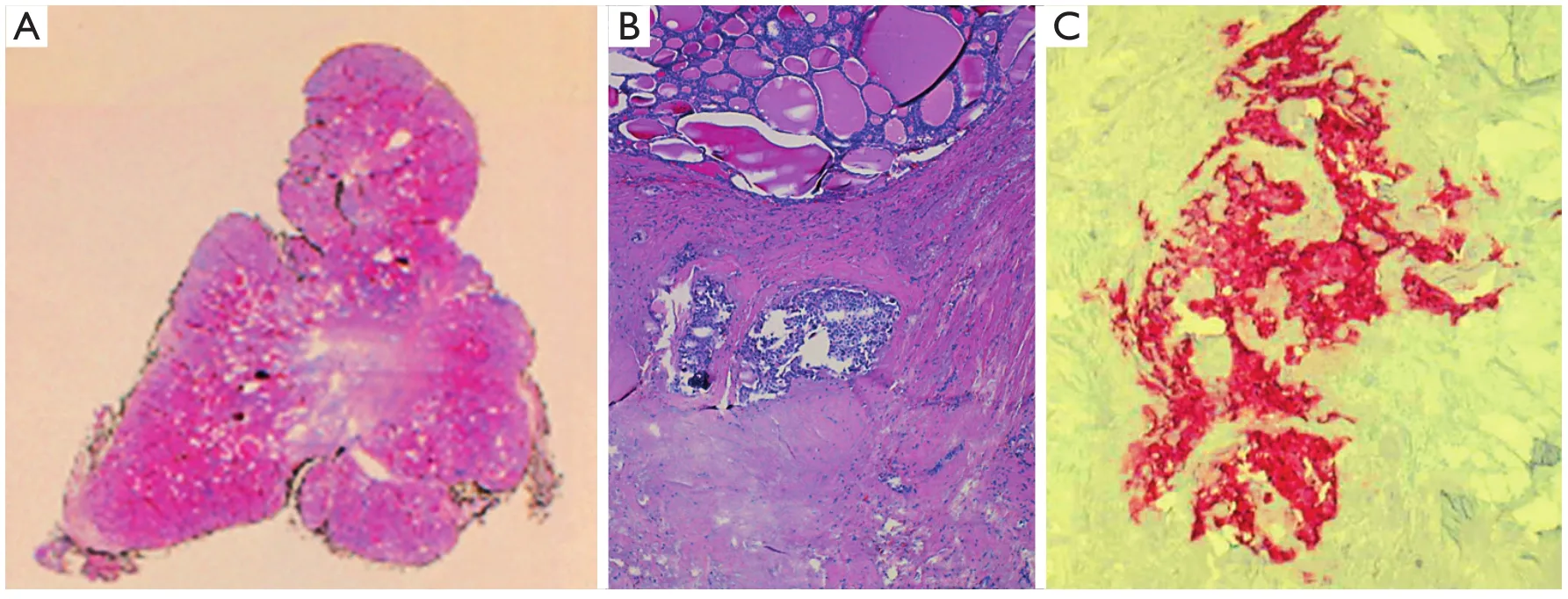
Figure 2 Medullary thyroid carcinoma: histological findings.(A) Scan of original histological slide (H&E staining, pale central area corresponded to dense fibrous tissue infiltrated by tumor).(B) Two tumor nodules in dense fibrous tissue at the top of figure (H&E staining, 100×).(C) Tumor cells are positive for Calcitonin (Calcitonin staining, 200×).
After the surgery, hypertension remitted and urinary adrenal hormones reverted to normal levels.However, postoperative serum calcitonin level was high (426 pg/mL) and the stimulation test with calcium gluconate showed increased levels of calcitonin.Subsequent CT and bone scintigraphy with metaIodobenzylguanidine and gallium did not show any conclusive findings.The suspicion of the MEN2A syndrome was confirmed through evaluation of the RET gene.The results showed p.C634R mutation in exon 11 and polymorphisms in p.A432A, p.G691S, and p.S904S.
Discussion
The patient case can be explained by adrenergic-induced ACS triggered by pheochromocytoma and hyperthyroidism.The delayed diagnosis of pheochromocytoma could be due to the concomitant presence of hyperthyroidism.
Germline RET mutations are generally observed in 98% of patients with MEN2A22.In the present case, a mutation was found in the 634 codon of the exon 11 of the RET gene.RET mutations are mostly missense and located in the extracellular domain of RET (exons 10 and 11)23and are correlated with high frequency of pheochromocytoma (more than 50%)24.The risk of developing MTC depends on RET mutation and can be prevented with prophylactic thyroidectomy.As such,genetic analysis must be performed in patients with a family history of MEN2A to perform early diagnosis and preventive thyroidectomy and thus avoid cervical neck dissection.
The American Thyroid Association has graded risk development and surgical timing from D (higher risk) to A (lower risk).Mutations at codon 634 of the RET gene correspond to a C classification, which is associated with high risk of aggressive MTC25.The phrase is appropriate as it is now stated.
Primary hyperparathyroidism is a frequent complication of MEN2A syndromes23.In the present case, this diagnosis was ruled out because of biochemical assessment and surgery results.As parathormon levels were normal pre- and postoperatively,the parathyroid was explored during the surgery, which revealed normal macroscopic anatomy.
In conclusion, ACS was probably secondary to intense coronary vasospasm caused by underlying adrenergic stimulation by pheochromocytoma and hyperthyroidism, which are the first clinical presentations of the MEN2A syndrome.
Conflict of interest statement
No potential conflicts of interest are disclosed.
1.Pappachan JM, Raskauskiene D, Sriraman R, Edavalath M,Hanna FW.Diagnosis and management of pheochromocytoma: a practical guide to clinicians.Curr Hypertens Rep 2014;16:442.
2.Eisenhofer G, Peitzsch M.Laboratory evaluation of pheochromocytoma and paraganglioma.Clin Chem 2014;60:1486-1499.
3.Barski D.Management and follow up of extra-adrenal phaeochromocytoma.Cent European J Urol 2014;67:156-161.
4.Baudin E, Habra MA, Deschamps F, Cote G, Dumont F, Cabanillas M, et al.Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma.Eur J Endocrinol 2014;171:R111-R122.
5.Maher ER, Eng C.The pressure rises: update on the genetics of phaeochromocytoma.Hum Mol Genet 2002;11:2347-2354.
6.Thakker RV.Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4).Mol Cell Endocrinol 2014;386:2-15.
7.Galetta F, Franzoni F, Bernini G, Poupak F, Carpi A, Cini G, et al.Cardiovascular complications in patients with pheochromocytoma: a mini-review.Biomed Pharmacother 2010;64:505-509.
8.Zelinka T, Petrák O, Turková H, Holaj R, Strauch B, Kršek M, et al.High incidence of cardiovascular complications in pheochromocytoma.Horm Metab Res 2012;44:379-384.
9.Prejbisz A, Lenders JW, Eisenhofer G, Januszewicz A.Mortality associated with phaeochromocytoma.Horm Metab Res 2013;45:154-158.
10.Horton WB, Frey LM, Hawkins UA, Ahmad SR.Pheochromocytoma presenting as acute non-ST elevation myocardial infarction following elective hysterectomy.J Miss State Med Assoc 2015;56:4-6.
11.Yang TH, Tsai WC.Recurrence and metastasis of pheochromocytoma mimic acute ST-segment elevation myocardial infarction: a case report.Am J Emerg Med 2015;33:311.e3-311.e5.
12.Martinez-Quintana E, Jaimes-Vivas R, Cuba-Herrera J, Saiz-Udaeta B, Rodríguez-Gonzalez F, Martinez-Martin MS.Acute myocardial infarction secondary to catecholamine release owing to cocaine abuse and pheochromocytoma crisis.Int J Endocrinol Metab 2013;11:48-51.
13.Subramanyam S, Kreisberg RA.Pheochromocytoma: a cause of ST-segment elevation myocardial infarction, transient leftventricular dysfunction, and takotsubo cardiomyopathy.Endocr Pract 2012;18:e77-e80.
14.Yu R, Nissen NN, Bannykh SI.Cardiac complications as initial manifestation of pheochromocytoma: frequency, outcome, and predictors.Endocr Pract 2012;18:483-492.
15.Coroleu SF, Muñoz-Camacho JF, Fernández-Gómez C, Tarroch-Sarasa X.Acute myocardial infarction probably related to severe coronary vasospasm during pheochromocytoma crisis.Rev Esp Cardiol 2011;64:244-246.
16.Martin JF, Martin LN, Yugar-Toledo JC, Loureiro AA, Cury PM,Júnior HM.Coronary emergency and diabetes as manifestations of pheochromocytoma.Int J Cardiol 2010;139:e39-e41.
17.Tamdy A, Oukerraj L, Khatri D, Ait Bella S, Etalibi N, Fetouhi H,et al.Acute myocardial infarction revealing a pheochromocytoma:a case report.Ann Cardiol Angeiol (Paris) 2010;59:97-99.
18.Goh YS, Tong KL.Phaeochromocytoma the great mimicker: a case report.Ann Acad Med Singapore 2008;37:79-81.
19.Tournoux F, Bal L, Hamoudi N, Desmonts JM, Steg PG.Acute coronary syndromes and pheochromocytoma.Ann Cardiol Angeiol (Paris) 2004;53:273-275.
20.Bachurska S, Staykov D, Belovezhdov V, Sasano H, Gulinac M,Stefanov C, et al.Bilateral pheochromocytoma/intra-adrenal paraganglioma in von Hippel-Lindau patient causing acute myocardial infarction.Pol J Pathol 2014;65:78-82.
21.Beedupalli J, Akkus NI.Concealed pheochromocytoma presenting as recurrent acute coronary syndrome with STEMI: case report of a patient with hyperthyroidism.Herz 2014;39:476-480.
22.Yeganeh MZ, Sheikholeslami S, Hedayati M.RET proto oncogene mutation detection and medullary thyroid carcinoma prevention.Asian Pac J Cancer Prev 2015;16:2107-2117.
23.Krampitz GW, Norton JA.RET gene mutations (genotype and phenotype) of multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma.Cancer 2014;120:1920-1931.
24.Frank-Raue K, Rybicki LA, Erlic Z, Schweizer H, Winter A,Milos I, et al.Risk profiles and penetrance estimations in multiple endocrine neoplasia type 2A caused by germline RET mutations located in exon 10.Hum Mutat 2011;32:51-58.
25.American Thyroid Association Guidelines Task Force, Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, et al.Medullary thyroid cancer: management guidelines of the American Thyroid Association.Thyroid 2009;19:565-612.
杂志排行

Cancer Biology & Medicine的其它文章
- Quantitative proteomic analysis for high-throughput screening of differential glycoproteins in hepatocellular carcinoma serum
- Development of targeted therapies in treatment of glioblastoma
- Advances in immunotherapy for treatment of lung cancer
- Fueling the engine and releasing the break: combinational therapy of cancer vaccines and immune checkpoint inhibitors
- Reality of evidence-based practice in palliative care
- Single-cell analyses of circulating tumor cells