Is hyperexcitability really guilty in amyotrophic lateral sclerosis?
2015-12-15FelixLeroy,DanielZytnicki
Is hyperexcitability really guilty in amyotrophic lateral sclerosis?
Amyotrophic lateral sclerosis (ALS) is a lethal disorder characterized by the gradual degeneration of brainstem and spinal motoneurons as well as lateral cortico-spinal tracts. The onset generally occurs during the adult age except for some juvenile aggressive forms. Until recently, the vast majority of the cases (90%) were deemed sporadic. Mutations in the SOD1 gene have been for a long time the only ones reported in familiar forms of ALS. However, the recent implication of new genes of little known function cast a new view on this disease. C9ORF72, for example, is now recognized to account for 30% of the familial cases. Overall, ALS has been linked to 20 diff erent genes, many also associated with other degenerative diseases (frontotemporal dementia, Alzheimer or ataxia). Some of these genes are involved in RNA maturation (FUS, TARBD). Clinical observation of human patients and mouse models suggests that all motor pools and motoneurons are not equally aff ected (see Kanning et al., 2010). The disease usually starts in motor pools controlling the limbs or in the bulbar area before expanding to other motor pools, with the exception of a few resistant ones (Onuf’s and oculomotor nuclei). Within a vulnerable motor pool, motoneurons subtypes also exhibit diff erential vulnerability and follow an orderly degeneration; starting with the motoneurons innervating fast-contracting fatigable motor units (FF motoneurons) and followed by the ones innervating fast-contractile fatigue-resistant motor units (FR motoneurons). The motoneurons innervating slow motor units (S motoneurons) appear resistant to the disease. Although the mechanisms leading to the orderly degeneration are not known, many hypotheses have been raised (Ilieva et al., 2009).
Extrinsic and intrinsic causes leading to an abnormal calcium entrance in the motoneurons: It has been suggested that an abnormal calcium entrance in motoneurons might be toxic and trigger degeneration (Ilieva et al., 2009). Indeed, calcium concentration in the cytoplasm is kept low and tightly regulated in all cells (Figure 1). Interestingly, vulnerable fast motoneurons express lower amounts of cytosolic calcium buff ering proteins, which could make them more sensitive to the large and fast fl uxes of calcium they experience (Kanning et al., 2010). In addition, the progression of the disease in fast motoneurons correlates with a decrease in calreticulin, an endoplasmic reticulum calcium buff ering protein, as well as a corresponding increase in cytosolic calcium concentration (Kanning et al., 2010). In neurons, calcium can enter the cytoplasm through numerous calcium channels that can be sorted according to their activation mechanism: ligand-gated channels (AMPA, NMDA), voltage-activated channels (N/P-, L- or T-type channels) and second-messenger-activated channels (IP3, G-protein receptors, Figure 1). Electrophysiological work has aimed to investigate whether changes in synaptic inputs or intrinsic electrical cell properties lead to more calcium entrance. By defi nition, changes seen in the former are deemed as a hyperexcitation, while changes in the latter are referred to as a hyperexcitability (Figure 1). Hyperexcitation originates from a shift in the balance of excitation and inhibition towards more net excitation. This can occur via increased excitation, decreased inhibition, and also altered glial activity. On the other hand, hyperexcitability can stem from any intrinsic mechanisms resulting in increased fi ring: higher input resistance, lower rheobase, depolarized resting potential, and hyperpolarized spiking threshold. It can also stem directly from an increase in calcium channel activity.
Are vulnerable motoneurons hyperexcitable? Original studies on the SOD1 mutant mouse ALS models focused on electrical markers of hyperexcitability at diff erent pre-symptomatic stages. Motoneurons from mSOD1 embryos recorded in culture were found to be hyperexcitable (Kuo et al., 2005; van Zundert et al., 2008) in that they were recruited at lower current than WT motoneurons and their frequency of discharge (F) increased more with injected current (I) (higher F-I curve slope). Martin et al. (2013) found a similar result in an in vitro preparation of mSOD1 embryonic cord. In their preparation, shrinkage of the dendritic tree was associated with an increased input resistance. In mSOD1 neonates, hyperexcitability remained controversial until recently (see Leroy et al., 2014). The major caveat of the previous work on neonates was that the diff erential sensitivity of motoneurons subtypes to the disease was not taken into account. Leroy et al. (2014) established a link between discharge patterns and slow and fast motoneurons. Separate analysis of each subtypes allowed them to demonstrate that, surprisingly, the excitability of the vulnerable fast motoneurons was unchanged whereas the slow motoneurons became hyperexcitable. Hyperexcitability of the slow motoneurons arose from a decrease in their rheobase and a hyperpolarization of the spike threshold. Overall, studies at early stages, far from the disease onset, indicate that though motoneurons can display hyperexcitability, it is unlikely to be responsible for their later degeneration.
Until recently, the inability to record from mouse adult motoneurons prevented to look for hyperexcitability at the end of the pre-symptomatic phase. Delestree et al. (2014) recorded motoneurons in vivo in SOD1 G93A mice. At the population level, they found that every hallmark of excitability remained unchanged despite a decrease in input conductance. Some motoneurons, however, lost the ability to fi re repetitively. Using a diff erent preparation, Hadzipasic et al. (2014) recorded motoneurons in acute slices made from adult SOD1 G85R mice. They performed a cluster analysis to categorize their motoneurons into pseudo-physiological types. Regardless of the types considered, they observed no change in input resistance. Furthermore, the motoneurons supposedly innervating the fastest motor units exhibited hyperpolarized resting potentials, which could manifest as hypoexcitability. Overall, both studies in adults failed to report any signs of hyperexcitability whereas they saw diff erent signs of hypoexcitability. Rather than to record from adult mouse motoneurons, several studies used a preparation that diff erentiated fi broblasts from ALS patients into motoneurons (induced pluripotent stem cells or IPSC), therefore, specifically investigating cell-autonomous mechanisms of ALS. The fi rst two studies on IPSC derived either from mSOD1 (Wainger et al., 2014) or C9ORF72 (Sareen et al., 2013) patients brought seemingly contradictory results. After 4 weeks of diff erentiation, Wainger et al. (2014) noted that SOD1 patient-derived motoneurons fi red spontaneously more often, albeit exhibiting a lower input resistance. They linked the hyperexcitability to a decrease in delayed-rectifi er potassium currents (Kv7). In contrast, working on C9ORF72 patient-derived motoneurons, Sareen et al. (2013) diff erentiated their IPSC for 8 weeks. They observed a
lower frequency of fi ring upon current injection and increased KCNQ3 expression, which led them to conclude that the motoneurons were hypoexcitable. The expression of other genes involved in membrane excitability, such as DPP6 and 3 members of the cerebellin family of proteins involved in synapse formation (CBLN1-3), was also altered. Devlin et al. (2015) recently published a study reconciling the two previous IPSC studies. They derived IPSC from TARDBP and C9ORF72 ALS patients. In their case, the gain of the F-I curve initially increased leading to hyperexcitability as in Wainger et al. (2014). However, hyperexcitability was transient and was later followed by a decrease in the sodium and potassium currents generating the action potentials. As in the studies by Sareen et al. (2013) and Delestree et al. (2014), the cells capable of sustained fi ring progressively lose this ability. Studies in adult and IPSC confirmed that hyperexcitability is a transient, early phenomenon absent in adults. Therefore, if anything, adult motoneurons are hypoexcitable. The fact that, in early stages, only the resistant motoneurons display hyperexcitability makes it unlikely that an initial phase of hyperexcitability during early development could trigger the late degeneration. The reason(s) why the largest adult motoneurons lose the ability to fi re repetitively and whether this may lead to degeneration remains to be investigated.
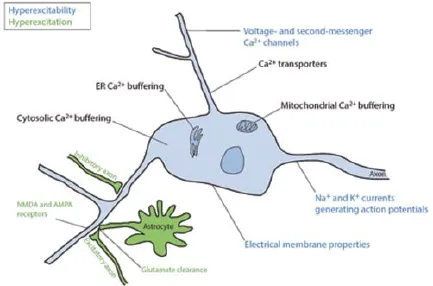
Figure 1 Mechanisms regulating the motoneuron spiking activity and cytosolic calcium concentration.
If hyperexcitability is not guilty, what about synaptic hyperexcitation? We know since Charcot’s work that the lateral cortico-spinal tract also degenerates during the ALS. The tract comprises axons from the motor cortex pyramidal neuron that control the excitation of local network upstream of the motoneurons at every spinal cord level. They might therefore indirectly trigger the hyperexcitation of the motoneurons. Indeed, transcranial magnetic studies in human suggest that the cortex become hyperexcitable in early stages of the disease (Vucic et al., 2008). In SOD1 G93A mice, Saba et al. (2015) found that the neurons in layer V of motor cortex were hyperexcitable due to a lower rheobase. In addition, they also observed a higher frequency of excitatory post-synaptic currents, larger dendritic arbor and increased expression of VGLUT2 in layer 5. This suggests that increased excitation could originate early on along the motor command chain and trickle down to the fi nal eff ectors, the motoneurons (Figure 1). In a mouse model of spinal muscular atrophy, another motor neuron disease, Mentis et al. (2011) showed that motoneuron hyperexcitability was not a primary disease mechanism but a homeostatic response to compensate for the decrease in proprioceptive inputs. However, due to the extensive pre-symptomatic period, it is more diffi cult to separate direct from compensatory mechanisms in ALS.
Dysfunctions in spinal interneurons and glial cells have also been observed in ALS (Pullen and Athanasiou, 2009; Martin and Chang, 2012; Wootz et al., 2013). A decrease in the amplitude of the glycinergic miniature currents in a cultured motoneuron preparation was reported as well as a global decrease in glycine synapses in slice (Martin and Chang, 2012; Wootz et al., 2013). Moreover, the ventral root initial response following dorsal root stimulation decreased (Bories et al., 2007) and cholinergic C-button synapses impinging on the motoneurons are enlarged (Pullen and Athanasiou, 2009). Finally, other cell types are involved in the degeneration as shown by co-cultures and chimeric mice experiments (see Ilieva et al., 2009). The EAAT2/GLT1 glutamate transporter is impaired in astrocytes, compromising glutamate clearance from the synaptic clefts (see Ilieva et al., 2009). Additionally, Calvo-Gallardo et al. (2015) found the SOD1 G93A chromaffi n cells to be hypoexcitable due to a decrease in nicotinic currents. Exactly how and which cells surrounding the motoneurons contribute to the disease is still unclear and more work needs to be undertaken to link this to altered excitability and to motoneuron degeneration.
Recent challenging observations by Saxena et al., (2013) suggest that hyperexcitation could in fact be benefi cial to motoneuron survival. Instead of assessing the putative changes in spinal cord activity, Saxena et al. (2013) altered activity throughout the central nervous system and observed the eff ect of this manipulation on motoneuron degeneration. Blocking of the serotonin, AMPA or NMDA receptors induced an increased accumulation of misfolded SOD1 and unfolded protein response resulting in an acceleration of the disease progression. Conversely, injecting mice with moderate doses of AMPA delayed the disease progression and extended the mutant mice lifespan (and not that of the wild-type). Though they show that a mild hyperexcitation of the spinal cord slows the disease progression, what would be the eff ect of a stronger hyperexcitation? Could a strong increase in excitation be detrimental while a lower one is benefi cial? More experiments are required to elucidate whether synaptic hyperexcitation occurs in ALS and how it specifi cally aff ects the vulnerable motoneurons.
To conclude, intrinsic hyperexcitability is unlikely to lead to motoneuron degeneration in ALS since vulnerable motoneurons are not hyperexcitable. In the juvenile mouse, only the resistant
motoneurons are hyperexcitable. Moreover, in the adult mouse, the vulnerable motoneurons tend to become hypoexcitable. In this scheme, higher excitability is associated with the resistant motoneurons, suggesting a benefi cial eff ect for hyperexcitability. However, for Roselli and Caroni (2015), hyperexcitability is merely a maladaptive consequence of early stress and calcium increase. Whether hyperexcitation, hyperexcitability or higher calcium concentration are responsible for the motoneuron degeneration in ALS is therefore still under investigation and after many years of intensive research on ALS, we are still looking for the mechanism(s) initiating the orderly degeneration. Surprisingly, Quinlan et al. (2015) recently reported that action potential-evoked calcium transients in juvenile motoneurons are overall smaller in SOD1 G93A animals. This might call for a reevaluation of the entire underlying hypothesis of higher calcium infl ux. At the motoneuron level, probing various sensitive cellular pathways such as the stress on the protein degradation complex or directly measuring calcium hotspots will help narrowing down how cells slowly progress toward degeneration. Targeted modifi cations (either at the cell or circuit level) of the diff erent components controlling the motoneuron calcium concentration are necessary to better understand the slow degeneration and complex compensatory mechanisms taking place throughout the disease.
Financial supports provided by the Agence Nationale pour la Recherche (HYPER-MND, ANR-2010-BLAN-1429-01), the NIHNINDS (R01NS077863), the Thierry Latran Fundation (OHEX Project) and Target ALS are gratefully acknowledged. Felix Leroy was recipient of a “Contrat Doctoral” from the Ecole Normale Supérieure, Cachan.
The authors would like to thank Philippe Ascher, Arjun Masurkar and Georges mentis and for their corrections and advice.
将10种酚类物质的标准混合溶液逐步稀释,配制成浓度分别为400,200,100,50,10 mg/L的混合标准溶液,在最佳色谱条件下进行检测,根据实验结果,绘制10种酚类物质的标准曲线,得出线性方程及相关系数,实验结果见表2。
Felix Leroy*,#, Daniel Zytnicki#
Centre de Neurophysique, Physiologie et Pathologie, UMR 8119, Université Paris Descartes, UMR 8119, 45 rue des
Saints-Pères, 752070 Paris Cedex 06, France
*Correspondence to: Felix Leroy, Ph.D., fl 2379@columbia.edu.
# These authors contributed equally to this work.
Accepted: 2015-05-29
Bories C, Amendola J, Lamotte d’Incamps B, Durand J (2007) Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci 25:451-459.
Calvo-Gallardo E, de Pascual R, Fernandez-Morales JC, Arranz-Tagarro JA, Maroto M, Nanclares C, Gandia L, de Diego AM, Padin JF, Garcia AG (2015) Depressed excitability and ion currents linked to slow exocytotic fusion pore in chromaffi n cells of the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Am J Physiol Cell Physiol 308:C1-19.
Delestree N, Manuel M, Iglesias C, Elbasiouny SM, Heckman CJ, Zytnicki D (2014) Adult spinal motoneurones are not hyperexcitable in a mouse model of inherited amyotrophic lateral sclerosis. J Physiol 592:1687-703.
Hadzipasic M, Tahvildari B, Nagy M, Bian M, Horwich AL, McCormick DA (2014) Selective degeneration of a physiological subtype of spinal motor neuron in mice with SOD1-linked ALS. Proc Natl Acad Sci U S A 111:16883-16888.
Ilieva H, Polymenidou M, Cleveland DW (2009) Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187:761-772.
Kanning KC, Kaplan A, Henderson CE (2010) Motor neuron diversity in development and disease. Ann Rev Neurosci 33:409-440.
Kuo JJ, Siddique T, Fu R, Heckman CJ (2005) Increased persistent Na(+) current and its eff ect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol 563:843-854.
Leroy F, Lamotte d’Incamps B, Imhoff-Manuel RD, Zytnicki D (2014) Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. ELife 3:e040406 doi:10.7554/eLife.040406.
Martin E, Cazenave W, Cattaert D, Branchereau P (2013) Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 54:116-126.
Martin LJ, Chang Q (2012) Inhibitory synaptic regulation of motoneurons: a new target of disease mechanisms in amyotrophic lateral sclerosis. Mol Neurobiol 45:30-42.
Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O’Donovan MJ (2011) Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69:453-467.
Pullen AH, Athanasiou D (2009) Increase in presynaptic territory of C-terminals on lumbar motoneurons of G93A SOD1 mice during disease progression. Eur J Neurosci 29:551-561.
Quinlan KA, Lamano JB, Samuels J, Heckman CJ (2015) Comparison of dendritic calcium transients in juvenile wild type and SOD1(G93A) mouse lumbar motoneurons. Front Cell Neurosci 9:139.
Roselli F, Caroni P (2015) From intrinsic fi ring properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron 85:901-910.
Saba L, Viscomi MT, Caioli S, Pignataro A, Bisicchia E, Pieri M, Molinari M, Ammassari-Teule M, Zona C (2015) Altered functionality, morphology, and vesicular glutamate transporter expression of cortical motor neurons from a presymptomatic mouse model of amyotrophic lateral sclerosis. Cereb Cortex doi: 10.1093/cercor/bhu317.
Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH (2013) Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5:208ra149.
Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P (2013) Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80:80-96.
van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH, Jr., Constantine-Paton M, Bellingham MC (2008) Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci 28:10864-10874.
Vucic S, Nicholson GA, Kiernan MC (2008) Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131:1540-1550.
Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH, Jr., Cudkowicz ME, Bean BP, Eggan K, Woolf CJ (2014) Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep 7:1-11.
Wootz H, Fitzsimons-Kantamneni E, Larhammar M, Rotterman TM, Enjin A, Patra K, Andre E, Van Zundert B, Kullander K, Alvarez FJ (2013) Alterations in the motor neuron-renshaw cell circuit in the Sod1(G93A) mouse model. J Comp Neurol 521:1449-1469.
10.4103/1673-5374.165308 http://www.nrronline.org/
Leroy F, Zytnicki D (2015) Is hyperexcitability really guilty in amyotrophic lateral sclerosis? Neural Regen Res 10(9):1413-1415.
猜你喜欢
杂志排行

中国神经再生研究(英文版)的其它文章
- Lactulose enhances neuroplasticity to improve cognitive function in early hepatic encephalopathy
- Elastic modulus aff ects the growth and diff erentiation of neural stem cells
- Optimal concentration and time window for proliferation and diff erentiation of neural stem cells from embryonic cerebral cortex: 5% oxygen preconditioning for 72 hours
- Stem Cell Ophthalmology Treatment Study (SCOTS) for retinal and optic nerve diseases: a case report of improvement in relapsing auto-immune optic neuropathy
- Repair of peripheral nerve defects with chemically extracted acellular nerve allografts loaded with neurotrophic factors-transfected bone marrow mesenchymal stem cells
- Polylactic-co-glycolic acid microspheres containing three neurotrophic factors promote sciatic nerve repair after injury