四(邻氟苄基)锡及一维链状三苄基锡二茂铁甲酸酯的合成、结构及性质
2015-12-05张复兴邝代治冯泳兰王剑秋庾江喜蒋伍玖朱小明
张复兴 邝代治 冯泳兰 王剑秋 庾江喜 蒋伍玖 朱小明
(衡阳师范学院化学与材料科学系,功能金属有机材料湖南省普通高等学校重点实验室,衡阳 421008)
四(邻氟苄基)锡及一维链状三苄基锡二茂铁甲酸酯的合成、结构及性质
张复兴*邝代治冯泳兰王剑秋庾江喜蒋伍玖朱小明
(衡阳师范学院化学与材料科学系,功能金属有机材料湖南省普通高等学校重点实验室,衡阳421008)
合成了四(邻氟苄基)锡(1)和一维链状三苄基锡二茂铁甲酸酯(2),经X射线衍射方法测定了化合物的晶体结构。化合物1属四方晶系,空间群I41/a,晶体学参数a=1.967 94(16)nm,b=1.967 94(16)nm,c=0.593 16(5)nm,V=2.297 2(3)nm3,Z=4,Dc= 1.605 g·cm-3,μ(Mo Kα)=11.58 cm-1,F(000)=1 112,R1=0.020 8,wR2=0.057 6。化合物2属单斜晶系,空间群为P21/n,晶体学参数:a=1.593 54(12)nm,b=1.007 23(8)nm,c=1.700 26(13)nm,β=91.001(10)°,V=2.728 6(4)nm3,Z=4,Dc=1.521 g·cm-3,μ(Mo Kα)=14.74 cm-1,F(000)=1 256,R1=0.038 4,wR2=0.095 2。晶体的中心锡原子分别呈四配位畸变四面体构型和五配位畸变三角双锥构型。对其结构进行量子化学从头计算,探讨了配合物的稳定性、分子轨道能量以及部分前沿分子轨道的组成特征。测定了配合物的热稳定性和体外抗癌活性。
四(邻氟苄基)锡;三苄基锡二茂铁甲酸酯;合成;结构;抗癌活性
烃基锡化合物由于具有结构的多样性、丰富的反应性、较强的生物活性和催化活性,多年来一直引起人们的兴趣[1-6]。烃基锡化合物中有四烃基锡、烃基卤化锡、烃基氧化锡、烃基锡羧酸酯配合物等多种类型。近年来人们合成了一系列具有结构特点和特殊性能的有机锡化合物[7-14],苄基锡化合物特别是取代苄基锡化合物的合成,更丰富了烃基锡化合物的内容。为了更进一步探索取代苄基锡类化合物的结构与性能的关系,本文合成了四(邻氟苄基)锡(1)和一维链状三苄基锡二茂铁甲酸酯(2),并通过元素分析、红外光谱对其结构进行了表征。用X-射线单晶衍射测定了该化合物的晶体结构,对其结构进行量子化学从头计算,探讨了配合物分子的稳定性、分子轨道能量以及一些前沿分子轨道的组成特征。测定了配合物的热稳定性和体外抗癌活性。
1 实验部分
1.1试剂和仪器
日本岛津IRPrestige-21红外光谱仪(4 000~400 cm-1,KBr),PE-2400元素分析仪,Bruker SMART APEXⅡ单晶衍射仪,spectramax M5连续光谱酶标测试仪,TG209F3热分析仪,X4数字显微熔点,温度计未经校正。所有试剂均为化学纯。
1.2化合物的合成
化合物1:在三颈瓶中加入14.5 g(0.1 mol)邻氟苄基氯,少量碘,60 mL正戊醇和40 mL邻二甲苯,回流搅拌下分批加入5.93 g(0.05 mol)经活化处理的锡粉和1.0 g镁条,快速搅拌回流反应6 h。趁热过滤出未反应的锡粉,滤液倒入100 mL 5%HCl溶液中,充分搅拌。冷却后分出水层,旋转蒸发部分溶剂后,放置析出白色固体。抽滤分离出固体,用少量冷无水乙醇洗涤固体,再用苯重结晶,得到无色晶体10.84 g,收率78.15%。熔点:92~94℃。红外光谱主要吸收峰:3 053(w),2 961(w),1 578(m),1 491 (s),14 541(m),519(m)cm-1。元素分析(C28H24F4Sn),计算值(%):C,60.57;H,4.33。实测值(%):C,60.22;H,4.24。
化合物2:在50 mL圆底烧瓶中,加入0.855 g(2 mmol)三苄基氯化锡、0.460 g(2 mmol)二茂铁甲酸、40 mL苯和2.5 mmol三乙胺,在电磁搅拌下加热回流反应5 h。趁热过滤除去不溶性固体,滤液旋转蒸发除去部分溶剂,放置析出棕色固体,用苯重结晶得棕红色晶体0.816 3 g,产率65.73%。熔点:257~259℃。红外光谱主要吸收峰:3 019(m),29 229(w),15 979(m),1 557(s),1 498(s),1 462(m),1 385(m),5 695(w),515(w)cm-1。元素分析(C32H30O2FeSn),计算值(%):C,61.89;H,4.87。实测值(%):C,61.78;H,4.82。
1.3晶体结构分析
分别选取大小为0.24mm×0.19 mm×0.17 mm(1)和0.29 mm×0.25 mm×0.23 mm(2)的晶体,在Bruker SMART APEXⅡCCD单晶衍射仪上,采用经石墨单色化的Mo Kα射线(λ=0.071 073 nm),于296(2) K,以φ~ω扫描方式收集数据。可观察衍射点分别为1 256个和5 185个(I>2σ(I))用于结构分析和精修。全部数据经Lp因子和经验吸收校正。晶体结构由直接法解出,非氢原子坐标通过数轮差值Fourier合成陆续确定,理论加氢法给出氢原子在晶胞中的位置坐标。对非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法修正。全部结构分析计算工作采用SHELX-97[15]程序系统完成。晶体学数据详见表1。
CCDC:1056036,1;1056037,2。

表1 化合物的晶体学数据Table1 Crystallographic data of the title complexes

续表1
1.4热稳定性和抗癌活性测定
利用TG209F3热分析仪,在空气氛中,升温速率为20℃·min-1的实验条件下测定配合物的热稳定性。
采用MTT法[16]检测化合物对人宫颈癌细胞(HeLa)的体外抗性肿瘤活性。将化合物用二甲基亚砜(DMSO)配成5.0 mg·mL-1的溶液,用RPMI-1640培养基分别稀释成5,10,25,50,100 μg·mL-1。取处于指数生长期的HeLa细胞悬液加至96孔板中(细胞浓度为50 000个·mL-1,100 μL每孔),于37℃、5%CO2恒温箱中培养16 h使细胞贴壁。去除上清,加100 μL不同浓度的上述化合物,每个浓度设 4个复孔,孵育24 h,弃去上清,每孔加入2.0 mg·mL-1的MTT溶液60 μL,继续培养3h,除上清液后,每孔加入150 μL二甲基亚砜,低速振荡10 min,使深蓝色结晶溶解,用酶标仪在490 nm波长处测其吸光度值。按如下公式:抑制率r=(OD对照组-OD测试组)/ OD对照组×100%,计算各组对癌细胞的抑制率。每组实验均重复3次,取其平均值。
2 结果与讨论
2.1晶体结构描述
化合物的主要键长和键角分别列于表2、3,化合物的分子结构见图1、图2。

表2 化合物1的主要键长(nm)和键角(°)Table2 Selected distances(nm)and angles(°)of complex 1

表3 化合物2的主要键长(nm)和键角(°)Table3 Selected distances(nm)and angles(°)of complex 2
化合物1:由分子结构图和结构参数可知,化合物中心锡原子与4个亚甲基碳原子相连形成四面体构型。4个Sn-C键键长相等,均为0.216 52(19) nm。锡原子和与之相连的碳原子之间所构成的键角中∠C(1i)-Sn(1)-C(1iii)与∠C(1ii)-Sn(1)-C(1)相等,为110.78(11)°,其余的4个键角相等,为108.82(6)°。分子中0.216 52 nm的Sn-C键键长,比四(对氟苄基)锡的Sn-C键平均键长(0.216 2 nm)[17]稍长,这是因为氟原子在邻位,使得中心锡原子周围的空间拥挤程度有所增加;而比三(邻氟苄基)氯化锡的Sn-C键平均键长(0.214 3 nm)[17]要长得多,是因为氯原子被体积大很多的邻氟苄基取代后,使得中心锡原子周围的空间拥挤程度大大增加。

图2 化合物2的分子结构图(椭球概率30%)Fig.2 Molecular structure of complex 2 with the ellipsoids drawn at the 30%probability level
化合物2:从分子结构图和结构参数可知,三苄基锡通过羧基桥联形成一维链状结构。中心锡原子分别与3个苄基的亚甲基碳、不同的二茂铁甲酸分子的2个羧基氧,形成五配位的三角双锥结构。3个苄基碳原子C(1)、C(8)、C(15)占据了三角双锥赤道平面的3个位置,2个氧原子O(1)、O(2i)则占据了赤道平面两侧的轴向位置。处于轴向位置的氧原子O(1)、O(2i)与处于赤道位置的碳原子的键角分别为:C(1)-Sn(1)-O(1)92.20(12)°、O(1)-Sn(1)-C(8)93.65(13)°、O(1) -Sn(1)-C(15)98.74(14)°、C(1)-Sn(1)-O(2i)79.74(12)°、C(8)-Sn(1)-O(2i)84.79(12)°、C(15)-Sn(1)-O(2i)90.42(13)°,都与90°有偏差,其偏差范围在8.74°~10.26°。处于轴向位置的原子的键角O(1)-Sn(1)-O(2i)169.97(9)°,比180°线性角小了10.03°。中心锡原子与处于赤道平面的3个碳原子间的夹角分别为:C(1)-Sn(1)-C(15) 118.82(17)°、C(1)-Sn(1)-C(8)116.66(16)°、C(15)-Sn(1)-C(8)122.33(15)°,3个角之和为357.81°,与360°有较大的偏差。由此可知化合物为畸变程度较大的三角双锥结构。
化合物2的结构中2个羧基氧原子以双齿配位分别与2个不同的锡原子成键,其键长分别为:Sn(1)-O(1)0.214 6 nm和Sn(1i)-O(2)0.243 0 nm,由此而形成了一维链状的分子结构,如图3所示。
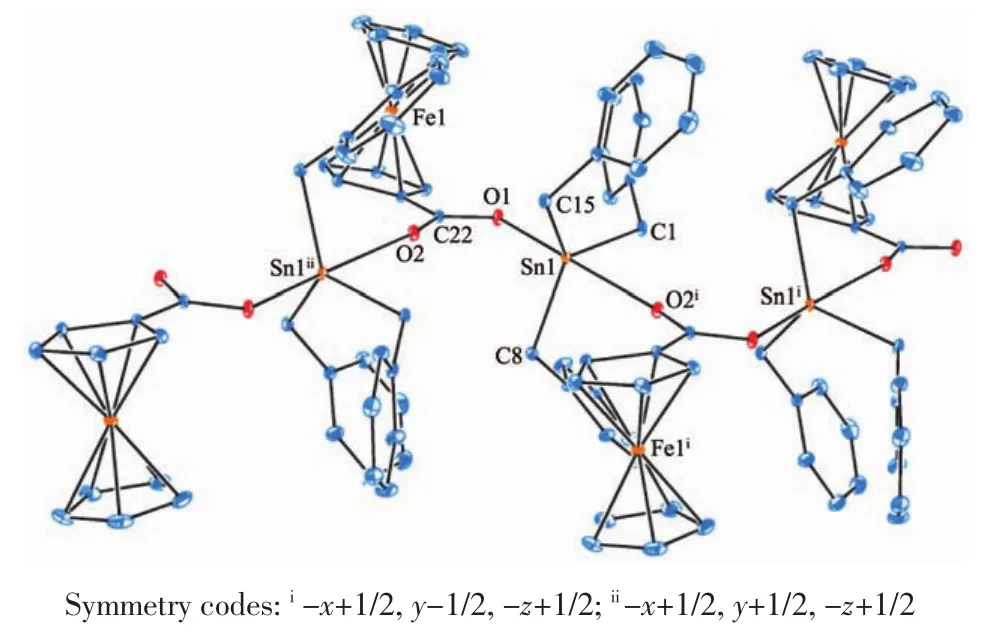
图3 化合物2的一维链状结构Fig.3 One-dimensional chain structure of complex 2
2.2量子化学研究
2.2.1分子的总能量和前沿分子轨道能量
根据晶体结构的原子坐标,运用Gaussian 03W程序和B3ylp/lanl2dz基组水平,计算得到分子的总能量和前沿分子轨道能量。
化合物1:ET=-373.141 486 9 a.u.,EHOMO=-0.384 88 a.u.,ELUMO=-0.314 06a.u.,ΔELUMO-HOMO=0.070 82 a.u.。从体系能量和前沿轨道的能量分析,化合物总能量较高,前沿占有轨道能量虽然低,但前沿未占轨道能量也较低,最高占据轨道与最低未占轨道的能量间隙ΔE很小,仅为0.070 82 a.u.,表明化合物分子结构稳定性有限,从氧化还原或电荷转移的角度分析,化合物较容易失去电子而被氧化。
化合物2:ET=-1 513.896 132 3 a.u.,EHOMO=-0.198 50 a.u,ELUMO=-0.054 05 a.u.,ΔELUMO-HOMO=0.144 5 a.u.。从体系能量和前沿轨道的能量分析,化合物总能量低,前沿占有轨道能量较低,表明化合物分子结构有一定的稳定性。但前沿未占轨道能量也较低,最高占据轨道与最低未占轨道的能量间隙ΔE较小,只有0.144 5 a.u,因此,从氧化还原或电荷转移的角度分析,化合物也较容易失去电子而被氧化。
2.2.2轨道成分分析
为探索化合物的电子结构与成键特征,对化合物分子轨道进行分析,用参与组合的各类原子轨道系数的平方和来表示该部分在分子轨道中的贡献,并经归一化。
化合物1:把化合物原子分为五部分:(a)锡原子Sn;(b)亚甲基碳原子C;(c)苯环碳原子C;(d)氟原子F;(e)氢原子H,前沿占有轨道和未占有轨道各取5个,计算结果见表4和图4。

图4 化合物1的前沿分子轨道示意图Fig.4 Schematic diagram of frontier MO for complex 1

表4 化合物1的分子轨道组成Table4 Some calculated frontier molecular orbitals composition of complex 1
表4和图4显示化合物1分子的成键特征:①前沿占有分子轨道中,苄基苯环碳原子,对分子轨道的贡献最大,达到了79.33%,并且在深层次轨道中均有较大的贡献,说明苯环具有良好的共轭离域性和稳定性。②前沿占有分子轨道中,锡原子和苄基亚甲基碳原子对分子轨道贡献都较小,分别为6.83%和6.26%,并且在深层次轨道中的贡献也都较小,说明Sn-C键强度弱,分子的稳定有一定的限度。从键长来看,化合物1的Sn-C键比三(邻氟苄基)氯化锡和四(对氟苄基)锡[17]的Sn-C键平均键长长,这与分子的稳定性相吻合。③比较HOMO与LUMO的各类原子轨道成份,可以看出,当电子从HOMO激发到LUMO轨道时,主要是邻氟苄基上的电子向锡原子转移。
化合物2:把化合物原子分为五部分:(a)二茂铁基碳原子和铁原子A;(b)羧基碳原子和氧原子B;(c)苄基碳原子C;(d)锡原子Sn;(e)氢原子H,前沿占有轨道和未占有轨道各取5个,计算结果见表5和图5。
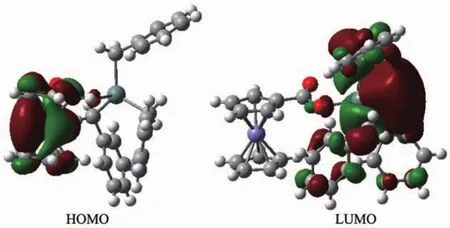
图5 化合物2的前沿分子轨道示意图Fig.5 Schematic diagram of frontier MO for complex 2
表5和图5显示化合物2分子的成键特征:①前沿占有分子轨道中,对分子轨道的贡献几乎来自二茂铁基,达到了99.24%,并且在深层次轨道中仍有较大的贡献,表明二茂铁基具有很好的稳定性。②前沿占有分子轨道中,与锡原子直接相连的基团对分子轨道贡献都很小,分别为羧基0.554 356%、苄基0.175 271%;而锡原子对分子轨道贡献则更小,只有0.011 323%;表明它们之间键的强度较弱,分子稳定性有限,这与从能量的角度分析的结果是一致的。③比较HOMO与LUMO的各类原子轨道成份,可以看出,当电子从HOMO激发到LUMO轨道时,主要是二茂铁基上的电子通过羧基向锡原子转移,羧基既是电子转移的桥梁,也是电子转移的受体。

表5 化合物2的分子轨道组成Table 5 Some calculated frontier molecular orbitals composition of complex 2
2.3热稳定性分析
化合物的热重曲线如图6、图7所示。

图6 化合物1的TG曲线Fig.6 TG curve of the complex 1
图6显示,随温度的升高,在初始阶段40~187℃,化合物1基本没有失重;在187~700℃区间,化合物出现明显失重,总计72.75%,对应于配合物分子失去4个邻氟苄基;最后稳定在约27.25%,残余物可被视为SnO2,与27.14%的计算值吻合;上述热分析结果表明化合物1在187℃之前是可以稳定存在的。

图7 化合物2的TG曲线Fig.7 TG curve of the complex 2
图7显示,随温度的升高,在初始阶段40~70℃,化合物2几乎没有任何失重;在70~530℃区间,化合物出现明显失重,总计63.65%,对应于配合物分子发生热分解失去有机基团;最后稳定在约36.34%,残余物可被假定为SnO2+Fe3O4,与36.69%的计算值吻合;上述热分析结果表明化合物2在70℃之前可以稳定存在,热稳定性比化合物1低。
2.4化合物的体外抗癌活性
不同浓度梯度下化合物对肝癌细胞的抑制率数值见表6。
由表可以看出2个化合物对HeLa细胞株增殖的抑制情况,化合物1在高浓度时对HeLa人宫颈癌细胞有一定的抑制能力,但随浓度降低其抑制率迅速变小,当浓度为5 μg·mL-1时,抑制率只有27.50%。而化合物2无论是在高浓度还是低浓度,都几乎没有抑制能力。

表6 不同浓度化合物对HeLa人宫颈癌细胞的抑制率Table 6 Growth inhibition rates for HeLa cell lines with title compounds at different concentrations
[1]Chandrasekhar V,Thirumoorthi R,Metre R K,et al.J. Organomet.Chem.,2011,696:600-606
[2]Effendy,Marchetti F,Marinelli A,et al.Inorg.Chim.Acta, 2011,366(1):388-393
[3]Munoz-Flores B M,Santillan R,Farfan N,et al.J.Organomet. Chem.,2014,769:64-71
[4]Hanif M,Hussain M,Ali S,et al.Polyhedron,2010,29:613-619
[5]ZHANG Xiao-Yan(张晓燕),YANG Guang(杨光),ZHANG Jun(张俊),et al.Chem.J.Chinsese Universities(高等学校化学学报),2010,31(6):1162-1166
[6]Siddiqi Z A,Shahid M,Kumar S,et al.J.Organomet.Chem., 2009,694:3768-3774
[7]Mahmudov K T,Guedes da Silva M F C,Kopylovich M N, et al.J.Organomet.Chem.,2014,760:67-73
[8]Ruan B F,Tian Y U,Zhou H P,et al.Inorg.Chim.Acta, 2011,365:302-308
[9]YINHan-Dong(尹汉东),WANG Chuan-Hua(王传华),WANG Yong(王勇),et al.Chinese J.Inorg.Chem.(无机化学学报), 2002,18(2):201-204
[10]YAN Wen-Hua(闫文华),KANG Wan-Li(康万利),LI Jin-Huan(李金环).Chinese J.Appl.Chem.(应用化学),2007,24 (6):660-664
[11]Shujha S,Shah A,Rehman Z U,et al.Eur.J.Med.Chem., 2010,45:2902-2911
[12]ZHANG Fu-Xing(张复兴),KUANG Dai-Zhi(邝代治),WANG Jian-Qiu(王剑秋),et al.Chinese J.Org.Chem.(有机化学), 2008,28(8):1457-1461
[13]KUANG Dai-Zhi(邝代治),JIANG Wu-Jiu(蒋伍玖),FENG Yong-Lan(冯泳兰),et al.Chinese J.Inorg.Chem.(无机化学学报),2012,27(10):1981-1986
[14]WANGJian-Qiu(王剑秋),ZHANGFu-Xing(张复兴),KUANG Dai-Zhi(邝代治),et al.Chinese J.Inorg.Chem.(无机化学学报),2015,27(31):237-2426
[15]Sheldrick G M.SHELXL-97,Program for Crystal Structure Refinement,University of Göttingen,Germany,1997.
[16]PENG Jun-Mei(彭俊梅),LI Wan(李婉),SHEN Kun(申坤), et al.Chem.J.Chinese Universities(高等学校化学学报), 2013,34(7):1646-1652
[17]ZHANGFu-Xing(张复兴),WANGJian-Qiu(王剑秋),KUANG Dai-Zhi(邝代治),et al.Chinese J.Org.Chem.(有机化学), 2003,23(4):368-371
Syntheses,Crystal Structures and Properties of the Tetra(o-fluorobenzyl)tin and the Tribenzyltin Ferrocenecarboxylate
ZHANG Fu-Xing*KUANG Dai-ZhiFENG Yong-LanWANG Jian-Qiu YU Jiang-XiJIANG Wu-JiuZHU Xiao-Ming
(Department of Chemistry and Material Science,Hengyang Normal University,Key Laboratory of Functional Organometallic Materials of Hengyang Normal University,College of Hunan Province,Hengyang,Hunan 421008,China)
The tetra(o-fluorobenzyl)tin(1)and the tribenzyltin ferrocenecarboxylate(2)have been synthesized.The crystal structures of the complexes were determined by X-ray diffraction.The crystal 1 belongs to tetragonal space group I41/a with a=1.967 94(16)nm,b=1.967 94(16)nm,c=0.593 16(5)nm,V=2.297 2(3)nm3,Z=4,Dc= 1.605 g·cm-3,μ(Mo Kα)=11.58 cm-1,F(000)=1 112,R1=0.020 8,wR2=0.057 6.The crystal 2 belongs to monoclinic space group P21/n with a=1.593 54(12)nm,b=1.007 23(8)nm,c=1.700 26(13)nm,β=91.001(10)°,V=2.728 6(4) nm3,Z=4,Dc=1.521 g·cm-3,μ(Mo Kα)=14.74 cm-1,F(000)=1 256,R1=0.038 4,wR2=0.095 2.The stabilities,some frontier molecular orbital energies and composition characteristics of some frontier molecular orbital of the complexes have been investigated by quantum chemistry calculation.Further more,thermal stability and anticancer activity of the complexes were tested.CCDC:1056036,1;1056037,2.
tetra(o-fluorobenzyl)tin;tribenzyltin ferrocenecarboxylate;synthesis;crystal structure
O614.43+2
A
1001-4861(2015)06-1194-07
10.11862/CJIC.2015.157
2015-03-11。收修改稿日期:2015-03-27。
湖南省自然科学基金(No.13JJ3112)、湖南省科技计划(No.2013TZ2025)、湖南省高校创新平台开放基金(No.13K105)、湖南省重点学科基金、湖南省高校科技创新团队支持计划和湖南省高校重点实验室开放基金资助。
*通讯联系人。E-mail:zfx8056@163.com;会员登记号:S060018907M。