Kimura 病 1 例报道并 81 例文献复习
2015-12-01胡袒伍骥吴迪郑超黄蓉蓉
胡袒 伍骥 吴迪 郑超 黄蓉蓉
Kimura 病 1 例报道并 81 例文献复习
胡袒 伍骥 吴迪 郑超 黄蓉蓉
目的 总结国内报道的 81 例 Kimura 病的临床资料,探讨 Kimura 病的流行病学及病理特点、临床表现、诊断与鉴别诊断和治疗及预后。方法 报告本院 2014 年收治的 1 例 Kimura 病,同时检索 2000 年1 月至 2014 年 12 月中文生物医学文献数据库、万方、中国知网、维普数据库;检索
为 Kimura、木村病、嗜酸性淋巴肉芽肿。结果 资料完整者 81 例 Kimura 病,加上本例共 82 例,其中男 63 例,女 19 例,男女比例为 3.3∶1,平均年龄 39 岁,平均病程 5.79 年。78.05% 的病例表现为头颈部皮下肿物,41.46% 同时伴有淋巴结病,单纯淋巴结肿大者 21.95%,21.95% 累及唾液腺,40.24% 伴有肿物局部症状,35.37% 伴有相关并发症,伴有外周血嗜酸性粒细胞计数增高占 85.37%,合并同时血清 IgE 升高占 42.68%。28.05% 的病例单纯手术治疗,18.29% 单纯激素治疗,45.12% 采取联合治疗。结论 Kimura 病是一种罕见的、慢性的、不明原因的、炎症性皮下软组织疾病,多见于亚洲青中年男性,常表现于头颈部皮下肿块,伴有局部淋巴结病和部分累及唾液腺,患者外周血及组织中嗜酸性粒细胞增多、血清 IgE 水平增高。手术、局部放射和类固醇激素治疗效果良好,但易复发。
嗜酸粒细胞增多性血管淋巴样增生;综述文献 ( 主题 );木村病
Kimura 病是一种少见的、慢性的、不明原因的、炎症性皮下软组织疾病,常位于头颈部,伴有局部淋巴结病,部分累及唾液腺,外周血及病损组织中常有嗜酸性粒细胞增多,血清 IgE 水平增高。现就我科收治的 1 例滑车上部淋巴结肿大的 Kimura病患者,结合 2000 年以来国内报道 81 例,对Kimura 病的诊断和治疗特点分析总结如下。
资料与方法
一、临床资料
患者,男,20 岁,主诉因发现左肘部肿物 1 年余,逐渐增大于 2014 年 10 月 21 日入院。患者入院前 1 年无意中发现左肘部肿物,约蚕豆大小,无疼痛,左肘关节无活动困难,未予诊治,肿物缓慢渐进性增大,现约核桃大小,轻压痛,无红肿、发热、疼痛及波动感,为求进一步诊治来我院门诊,遂以“左肘部肿物,性质待查”收入我科。病程中无发热、盗汗、乏力、体重下降及关节红肿。查体:36 ℃,脉搏:72 次 / min ,呼吸:20 次 / min,血压:100 / 65 mm Hg。左肘关节滑车上可见一肿物,大小约 2 cm×3 cm,位于肌层,质地软,活动度可,轻压痛,叩击肿物时无手指麻木,肿物边界欠清楚,与皮肤、基底粘连。四肢各关节活动正常,心肺腹部查体未见明显异常。入院后查血常规、尿常规、便常规+便潜血、凝血常规未见异常,生化示:血清磷 1.7 mmol / L↑( 0.5~1.6 ) mmol / L、血尿酸 538 μmol / L↑ ( 108~420 ) μmol / L。手术感染八项示:乙型肝炎表面抗体 151.35 mIU / ml↑阳性 ( 0~10 mIU / ml,阴性 )。血型示:正定 ( AB 抗原卡氏 ) AB 型、Rh 血型 ( D 抗原 ) 阳性,心电图、胸片及左肘部正侧位片未见明显异常 ( 图 1 )。
图 1 左肘关节正侧位:骨质及软组织未见明显异常Fig.1 Anteroposterior and lateral plain radiographs of left elbow joint showed no obvious abnormalities in bone and soft tissues
左肘部彩超提示:左侧肘关节内侧皮下低回声结节,多考虑增大淋巴结。患者于 2014 年 10 月24 日在局麻加强化下行左肘部肿物切除活检术,术中见:左肘部滑车上皮下肿物,大小约 1 cm×2 cm大小,质软,无包膜,与周围组织粘连,边界欠清楚,活动度一般。病理回报:( 左肘肿物 ) 淋巴组织反应性增生伴生发中心扩大 ( 图 2 ) 及嗜酸性粒细胞弥漫浸润 ( 图 3 ),副皮质区血管增生,局灶伴管壁玻璃样变 ( 图 4 ),符合 Kimura 病,免疫组化:CD20 (+) ( 图 5 )、CD3 (+)、CD21 (+)、Ki-67 ( 正常分布 )、Bcl-2 (+) ( 图 6 )。术后明确诊断为 Kimura 病。住院至切口愈合并拆线后出院。出院后准备予以口服小剂量激素治疗。目前随访 2 个月,患者来院复查见左肘部切口处 3 cm×1 cm 瘢痕样组织,凸向皮肤表面,伴有瘙痒 ( 图 7 ),复查血常规未见明显异常,建议进一步随访。
二、文献回顾
1. 文献检索
检索 2000 年 1 月至 2014 年 12 月,中文生物医学文献数据库、万方、中国知网、维普数据库,检索关键词为 Kimura、木村病、嗜酸性淋巴肉芽肿。共检索到有明确病理依据的 116 例报道,取其中资料较完整的 81 例,加上本例共 82 例,进行分析。
2. 病例分析
( 1 ) 一般资料:82 例均经病理检查 ( 或活检 )确诊为 Kimura 病,其中女 19 例,男 63 例 ( 儿童2 例 ),男女比例为 3.32∶1,年龄为 2~74 岁,平均 39 岁 ( 表 1 )。本组病程 1 周~25 年,平均5.79 年,其中 8 例病程超过 20 年,13 例病程 10~20 年。
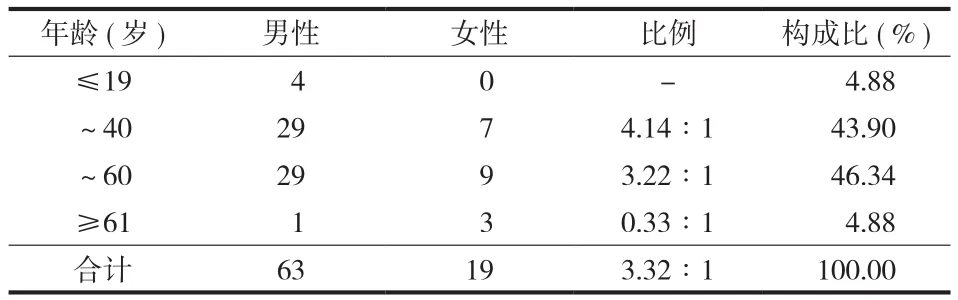
表 1 患者性别和年龄分布Tab.1
( 2 ) 临床表现:首发症状为无痛性肿物 75 例、肾脏疾病 4 例、皮疹 3 例;其中表现为头颈部皮下肿物 43 例,躯干四肢皮下肿物 9 例,全身多处散发皮下肿物 12 例,单纯淋巴结肿大 18 例 ( 颈部2 例、腋窝 2 例、腹股沟 1 例、滑车上 2 例、合并两处以上 11 例、累及纵隔淋巴结 2 例 )。同时伴有淋巴结病 34 例,累及腮腺 10 例、颌下腺 8 例,伴有局部瘙痒 21 例、局部压痛 3 例、局部皮肤破溃3 例、局部色素沉着 4 例,2 例伴有发热;13 例合并有肾脏疾病,9 例伴合并有皮疹,3 例伴合并有哮喘,1 例合并心、肺、肾脏多器官疾病,1 例合并分泌性中耳炎,1 例合并皮疹、哮喘、肾损害、血栓闭塞性脉管炎及下肢深静脉血栓,1 例伴发皮肤T 细胞淋巴瘤 ( 表 2、3 )。皮下肿物直径 0.5~20 cm,平均为 4 cm,单发病灶 32 例,多发病灶 50 例。
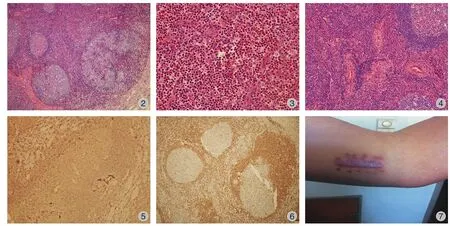

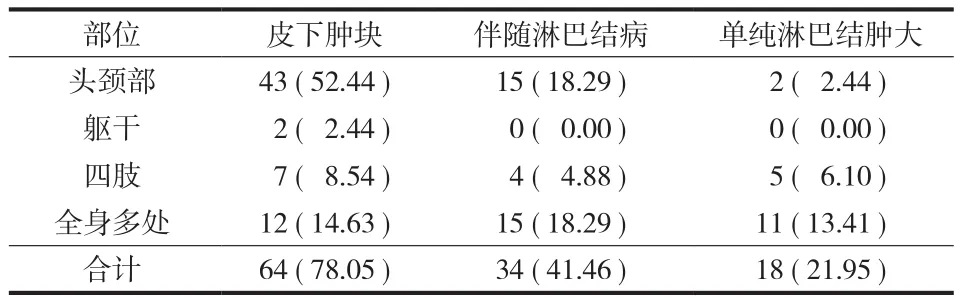
表 2 82例患者的主要临床表现 [ 例,百分比 ( % ) ]Tab.2 Main clinical manifestations of 82 cases ( case, ratio % )
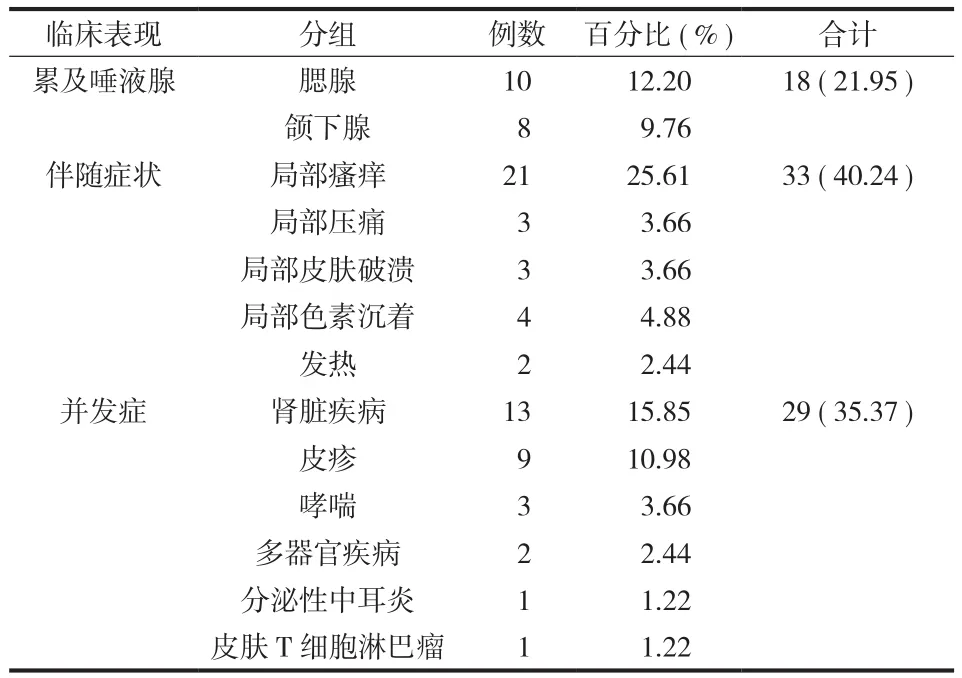
表 3 本组病例其它可见临床表现Tab.3 The other clinical manifestations in this group cases
( 3 ) 实验室检查:外周血嗜酸性粒细胞计数增高 70 例,分类计数为 0.06~0.72,平均 0.34± 0.27,绝对计数为 ( 0.7~16.0 ) ×109/ L,平均( 4.8±4.2 )×109/ L,正常 12 例,合并同时血清IgE 升高 35 例 ( 正常<113 IU / mL ),单纯 IgE 升高2 例,两者都正常 2 例,未测 IgE 水平者有 43 例;本组 82 例中,43 例检测了红细胞沉降率,有 5 例升高,平均为 29 mm / h。34 例患者尿蛋白阳性,有10 例达到诊断肾病综合征标准,23 例检测血清补体无异常。本组 82 例 85.37% 伴有外周血嗜酸性粒细胞计数增高,共计 70 例,45.12% 血清 IgE 升高,如同时存在皮下软组织肿物或淋巴结病,对早期诊断该病具有重要意义。
( 4 ) 诊断及病理表现:64 例手术切除肿物者,18 例淋巴结穿刺活检者,病理均证实为 Kimura 病,所有病例均符合 Kimura 病特点,即病变组织中广泛的淋巴滤泡样结构形成,滤泡间伴有大量嗜酸性细胞浸润、毛细血管增生,30 例具有淋巴滤泡间区、被膜、甚至被膜外脂肪组织内有大量成熟嗜酸性粒细胞浸润及嗜酸性微脓肿形成的特征性改变。本组病例中 12 例进行肾脏穿刺活检,证实为 7 例系膜增生性肾小球肾炎、3 例微小病变性肾炎、1 例膜性肾病、1 例 IgA 肾病,1 例 Kimura 病 38 年后取皮下增生结节病理证实为皮肤 T 细胞淋巴瘤。本组病例具有淋巴滤泡样结构形成、大量嗜酸性细胞浸润、毛细血管增生恒定性特征,66.67% 病例具有特征性嗜酸性微脓肿形成,有助于病理诊断 Kimura 病。
( 5 ) 治疗及预后:单纯手术治疗者 23 例,单纯激素治疗者 15 例,单纯放疗者 5 例,单纯免疫抑制剂治疗者 2 例,以上联合治疗者 37 例 ( 其中手术联合激素治疗者 12 例,其它为手术、放疗、细胞毒性药物、免疫抑制剂相互或多种联合 ),本组病例中有详细治疗经过并获随访者 15 例,随访 2 个月~5 年,其中 1 例治疗后 2 年死于感染性休克,余均未复发,截稿时仍在进一步随访中 30 例,未报道随访者 37 例,15 例获随访者复发者 9 例。
讨 论
Kimura 病,又名“嗜酸性淋巴肉芽肿”,是一种少见的、良性的、不明原因的、慢性炎症性的皮下软组织疾病[1],该病由我国学者金显宅[2]等 1938年首先以“嗜酸粒细胞增生性淋巴肉芽肿”描述报道,1948 年日本学者 Kimura 等[3]以“不寻常性淋巴组织增生性肉芽肿”并用英文形式报道,同时首次提出可能并发肾脏损害,Iizuka 等[4]于 1959 年探讨了该病的临床演变过程及病理特征并建议使用“Kimura 病”一名,自此,该病以“Kimura 病著称。目前,全世界关于该病的报道约 400 余例,主要集中于个案报道[1,5]。
一、流行病学及病因病理
Kimura 病的发病有明显的地域性,多发生在中国、日本及东南亚,但在欧洲和美国偶有散发病例报道,多见于亚洲人、偶见于白种人、罕见于非洲人。该病多发生于青年男性,高峰年龄为 20~40 岁,男女发病比例为 3.5~7.0∶1[6-8]。傅君芬[9]等回顾性分析中国 29 儿童病例报道得出:儿童男女发病比例为 28∶1,发病年龄 2.7~12 岁,平均12 岁,其临床表现与成人相似[10]。尽管存在大量关于 Kimura 病发病机理的理论,但病因目前仍不明确,它可能与异常自身免疫反应、过敏反应、肿瘤、感染、寄生虫、创伤、蚊虫叮咬等有关[2-3,11],相关文献报道患者外周血中 IL-4、IL-5、IL-13、TNF-α、GM-CSF、IgE 增多,病损组织中大量嗜酸性粒细胞、混有肥大细胞及浆细胞浸润,所以Kimura 病更多的被认为是 Th2 免疫调节紊乱 ( 导致上述细胞因子过度产生,从而促使 IgE 及嗜酸性粒细胞增殖 ) 或 IgE 介导的 I 型变态反应性免疫疾病[6,11]。Hui 等 1989 年将 Kimura 病组织病理特征划分为恒定的、常见的、罕见的特征,恒定的特征包括:发育良好的淋巴滤泡结构完好、滤泡增生伴反应性生发中心、嗜酸性细胞浸润、毛细血管后微静脉增生,常见的特征由生发中心蛋白样沉积、血管化、坏死,存在分散的多核的树突状细胞 ( 沃-芬型 ),嗜酸性粒细胞聚集、微脓肿形成,滤泡溶解,基质及静脉周围硬化组成,罕见生发中心进行性转化变异的特征,免疫组化显示 IgE 在生发中心呈特征性网状型,并由非-脱颗粒的肥大细胞包覆[7,12-13]。本组 82 例中,男女比例为 3.3∶1,有2 例儿童病例,病理证实具有典型 Kimura 病病理特点,这与国外及国内其它相关研究报道一致,本组病例集中于青中年男性,男性明显高于女性,可见儿童散发病例。平均病程 5.79 年,这表明该病病程缓慢,呈良性发展,预示预后较好。
二、临床表现
Kimura 病典型临床表现为位于头颈部的无痛性皮下软组织肿块,常多发、位置较深、边界不清、生长缓慢,类似良性肿瘤,常伴有局部淋巴结病 ( 主要是头颈部,偶见腋窝、腹股沟及滑车上 )和 ( 或 ) 累及唾液腺 ( 主要是腮腺、下颌下腺及小唾液腺 ),也有报道肿物位于耳廓、头皮、眼睑、眶周、泪腺、口腔、神经、输精管、鼻窦等不寻常部位[12,14-15],偶有肿块表面皮肤瘙痒、色素沉着、明显压痛、局部皮肤破溃。通常不伴有诸如发热、咳嗽、体重减轻等全身症状。该病具有特征性的三联征:无痛性单侧的头颈部皮下肿块;外周血及组织中嗜酸性粒细胞增多;血清 IgE 水平显著增高[16]。Kimura 病常合并肾脏疾病,12%~16% 为肾病综合征,可同时发生或先于 Kimura 病[17],Yadla 等[18]报道 1 例因肾脏疾病做血液透析后 1 年才出现右滑车上皮下肿物及外周血嗜酸性粒细胞增多。该病合并支气管哮喘[19]、嗜酸细胞性肺疾病[14]、雷诺现象[20]、湿疹[21]、荨麻疹、溃疡性结肠炎[21]、主动脉炎综合征[22]、肢体坏死及多发性单神经炎[23]、巨细胞动脉炎[1]、动静脉血栓形成[24]等并发症已有相关文献报道。Ishii[25]回顾性分析日本的 429 例后,发现 14 例伴有肾炎综合征、5 例伴有支气管哮喘、3 例伴有过敏性鼻炎、2 例伴有特应性皮炎、5 例伴有荨麻疹,他认为 KD 与免疫反应有关。本组病例中大多数主要表现为头颈部皮下肿物,常多发生在头颈部,呈多发性,41.46% 同时伴有淋巴结病,21.95% 累及唾液腺,40.24% 伴有肿物局部症状,35.37% 伴有相关并发症,伴有外周血嗜酸性粒细胞计数增高占 85.37%,合并同时血清 IgE 升高占 42.68%,这与国内其它及国外文献报道相符合。Kimura 病仅仅表现为滑车上淋巴结肿大鲜有报道,特别是同时嗜酸性粒细胞计数正常,我院收治的 1 例滑车上淋巴结肿大 Kimura 病患者,加上本组82 例中另外 1 例,共 2 例 ( 约 2.44% )。
三、诊断与鉴别诊断
目前,Kimura 病没有统一的诊断标准,结合临床表现及实验室检查可考虑此病,但最终确诊依靠活组织病理,其 CT 或 MRI 没有特征性表现[26-27]。临床上 Kimura 病常需与血管淋巴样组织增生伴嗜酸性粒细胞增多 ( angiolymphoid hyperplasia with eosinophilia,ALHE )、霍奇金淋巴瘤、嗜酸性肉芽肿、过敏性肉芽肿、寄生性淋巴结炎相鉴别,因为它们都可表现为淋巴结病或嗜酸性粒细胞增多,通过临床表现、实验室及病理检查不难作出鉴别诊断,但最重要的该病需与血管淋巴组织增生伴嗜酸性粒细胞浸润相鉴别,因为两者极其相似以至曾被认为是同一疾病。其鉴别要点为:( 1 ) 两者通常都表现为头颈部软组织肿物,但 ALHE 大部分位于表皮或皮下表浅的丘疹样结节,伴有出血、瘙痒及肿瘤性生长,常不累及淋巴结及唾液腺,血清中 IgE及嗜酸性粒细胞正常,而 Kimura 病刚好相反;( 2 ) ALHE 好发于中年女性,以西方人多见;Kimura 病则多见于年轻亚洲男性;( 3 ) 两者病理特征都是淋巴细胞浸润增生 ( Kimura 病为主 )、嗜酸性粒细胞浸润增多、生发中心形成、血管增生 ( ALHE 为主 ),但ALHE 血管内皮细胞核呈不同大小、形态各异并常有含铁血黄素沉积,且内皮细胞不止一层,异型性明显,无玻璃样变,而 Kimura 病为薄壁血管样增生,常为一层扁平内皮细胞,有明显玻璃样变,这些病理特征表明前者是肿瘤性起源,也称为上皮样血管瘤,而后者是免疫性疾病[6,14,19]。值得注意的是,本组病例中多数最初被诊断为淋巴结炎、淋巴结结核、肿瘤伴淋巴结转移,从而给予抗炎、抗结核、抗肿瘤等治疗,最后才经病理证实为 Kimura 病,说明该病早期诊断很困难,很多医生对该病缺乏认识,从而导致误诊误治而延误诊治。对于皮下肿块或淋巴结肿大患者,如果同时外周血嗜酸性粒细胞计数和血清 IgE 升高,需有足够证据来排除该病。
四、治疗及预后
目前对于 Kimura 病的最佳治疗方案存在争议,不同治疗方案有各自的优缺点,主要包括:手术切除、局部放疗、药物治疗 ( 局部或全身的甾体激素治疗、非甾体抗炎药、细胞毒性药物、维甲酸、环孢素、硫唑嘌呤、伊马替尼、配妥西菲林、奥马佐单抗、免疫球蛋白等 ),治疗方案的选择因人而异,但所有治疗方案都不可避免复发 ( 15%~80% )[28-29],邓维叶等[5]对 40 例 Kimura 病相关性分析得出增殖相关抗原 Ki-67 的表达与 Kimura 病的复发相关,可作为预测 Kimura 病患者复发的一个有效观察指标。当病变既没有引起症状也没有导致局部外观改变时,一般选择定期复查及随访;针对局部病变,手术切除病灶是第一选择,但是由于该病浸润性、无包膜及边界不清的特点,手术完全切除很困难,术后复发率较高,对于不完全切除或复发的病例,可辅助使用皮质激素或免疫抑制剂或局部放疗[30];局部放疗 ( 20~45 Gy,2~3 周 ) 对 90% 的局部病变有效,特别是多发、复发难治性病例,但不适合儿童[14]。全身或病灶内用泼尼松龙皮质激素能有效的减小病灶,特别是伴有肾病综合征者,但当泼尼松龙减量或停用时,该病又易复发。Abbas等[31]报道用光动力疗法能有效减缓疾病的进展并减少局部形变。Nonaka 等[32]研究证实抗 -IgE 抗体奥马佐单抗能降低外周嗜酸性粒细胞,减少炎症细胞浸润,从而缩小局部病变,可能成为一种潜在重要的治疗方法。尽管 Kimura 病局部复发、治疗效果波动,但是其总体预后良好,目前没有恶变病例报道[33],但 Akhavan 等[34]曾报道 1 例 Kimura 病合并同侧肾癌的患者,同时本组病例中陈辉树等[35]也报道 1 例 Kimura 病 38 年伴发皮肤 T 细胞淋巴瘤,提示 Kimura 病可能存在恶性转化。总之,治疗的目的在于保存功能和改善美观,防止复发。
[1] Chang AR, Kim K, Kim HJ, et al. Outcomes of Kimura's disease after radiotherapy or nonradiotherapeutic treatment modalities. Int J Radiat Oncol BiolPhys, 2006, 65(4): 1233-1239.
[2] Kim H, Szeto C. Eosinophilic hyperplastic lymphogranuloma, Comparison with Mikulicz's disease. Chin Med J, 1937, 23(69):700.
[3] Kimura T, Yoshimura S, Ishikawa E. On the unusual granulation combined with hyperplastic changes of lymphatic tissue. Trans Soc Pathol Jpn, 1948, 37(2):179-180.
[4] Iizuka S. Eosinophilic lymphadenitis and eosinophilic lymphoid granuloma: A proposal of the new concept of the disease ofthe lymph node and its surrounding tissue. Nihon Univ Med J, 1959, 18(1959):900-908.
[5] 邓维叶, 高云飞, 陈艳峰, 等. Ki-67在KD患者中的表达和复发相关因素分析. 中山大学学报(医学科学版), 2014, 4(35): 584-588.
[6] Meningaud JP, Pitak-Arnnop P, Fouret P, et al. Kimura's disease of the parotid region: Report of 2 cases and review of the literature. J Oral Maxillofac Surg, 2007, 65(1):134-140.
[7] Sah P, Kamath A, Aramanadka C, et al. Kimura's disease-An unusual presentation involving subcutaneous tissue, parotid gland and lymph node. J Oral Maxillofac Pathol, 2013, 17(3):455-459.
[8] Savage NW, Boras VV. Unilateral intraparotid swelling: a case report of Kimura's disease and review of differential diagnosis. Case Rep Otolaryngol, 2013, 2013:795921.
[9] Xu X, Fu J, Fang Y, et al. Kimura disease in children: a case report and a summary of the literature in Chinese. J Pediatr Hematol Oncol, 2011, 33(4):306-311.
[10] Hosoki K, Hirayama M, Kephart GM, et al. Elevated numbers of cells producing interleukin-5 and interleukin-10 in a boy with Kimura disease. Int Arch Allergy Immunol, 2012, 158(Suppl 1):S70-74.
[11] Chen H, Thompson LD, Aguilera NS, et al. Kimura disease: A clinicopathologic study of 21 cases. Am J Surg Pathol, 2004, 28(4):505-513.
[12] Kar IB, Sethi AK. Kimura's Disease: Report of a case and review of literature. J Maxillofac Oral Surg, 2013, 12(1): 109-112.
[13] Hui PK, Chan JK, Ng CS, et al. Lymphadenopathy of Kimura's disease. Am J Surg Pathol, 1989, 13(3):177-186.
[14] Ceçen E, Kaçar-Döger F, Etensel B. An extremely rare cause of generalized lymphadenopathy in children: Kimura's disease. Turk J Pediatr, 2010, 52(5):534-537.
[15] Saxe N, Kahn LB. Angiolymphoid hyperplasia with eosinophilia: report of 3 cases. S Afr Med J, 1977, 52(11):454-457.
[16] Mrówka-Kata K, Kata D, Kyrcz-Krzemień S, et al. Kikuchi-Fujimoto and Kimura diseases: the selected, rare causes of neck lymphadenopathy. Eur Arch Otorhinolaryngol, 2010, 267(1): 5-11.
[17] O'Malley DP, Grimm KE. Reactive lymphadenopathies that mimic lymphoma: entities of unknown etiology. Semin Diagn Pathol, 2013, 30(2):137-145.
[18] Yadla M, Sriramnaveen P, Sivakumar V, et al. Epitrochlear mass in a patient on maintenance hemodialysis--Kimura disease. Hemodial Int, 2012, 16(4):568-570.
[19] Moran CA, Suster S. Angiolymphoid hyperplasia with eosinophilia (epithelioid hemangioma) of the lung: a clinicopathologic and immunohistochemical study of two cases. Am J Clin Pathol, 2005, 123(5):762-765.
[20] Green SE, Linton SM, Harding JR, et al. Raynaud's phenomenon associated with Kimura's disease. Rheumatology (Oxford), 2005, 44(4):559-561.
[21] Khoo BP, Chan R. Kimura disease: 2 case reports and a literature review. Cutis, 2002, 70(1):57-61.
[22] Horie S, Ishiyama T, Sugimoto M, et al. Eosinophilic lymphofolliculosis (Kimura's disease) complicated with aortitis syndrome. Rinsho Ketsueki, 1989, 30(3):396-369.
[23] Eguia B, Bachmeyer C, Charlotte F, et al. Kimura disease with necrosis of the limbs and mononeuritis multiplex. Clin Exp Dermatol, 2011, 36(3):329-331.
[24] 韦秀宁, 郑东辉, 莫颖倩, 等. KD合并肾损害、支气管哮喘、血栓闭塞性脉管炎及深静脉血栓一例. 中华风湿病学杂志, 2014, 18(6):431-432.
[25] Ishii M. Kimura's disease: a review of 429 cases and four new cases. Oto-Rhino-Laryngology Tokyo, 1982, 25(4):407-416.
[26] Uysal IO, Eryilmaz MA, Salk I, et al. Kimura disease in the parotid gland. J Craniofac Surg, 2011, 22(1):337-338.
[27] Park SW, Kim HJ, Sung KJ, et al. Kimura disease: CT and MR imaging findings. AJNR Am J Neuroradiol, 2012, 33(4): 78478-8.
[28] Praeger AJ, Tsui A, Hardy TG. Kimura disease: rare cause of a slowly progressive orbital mass. Clin Experiment Ophthalmol, 2014, 42(4):385-387.
[29] Kim MB, Shin DH, Seo SH. Juvenile temporal arteritis with perifollicular lymphoid proliferation resembling Kimura disease. Report of a case. Int J Dermatol, 2011, 50(1):70-73.
[30] Kapoor NS, O'Neill JP, Katabi N, et al. Kimura disease: diagnostic challenges and clinical management. Am J Otolaryngol, 2012, 33(2):259-262.
[31] Abbas S, Jerjes W, Upile T, et al. Treatment of Kimura disease with photodynamic therapy: a case study. Photodiagnosis Photodyn Ther, 2012, 9(1):83-86.
[32] Nonaka M, Sakitani E, Yoshihara T. Anti-IgE therapy to Kimura's disease: a pilot study. Auris Nasus Larynx, 2014, 41(4):384-388.
[33] Nag VK, Nandan D, Bhardwaj M. Kimura's disease presenting with inguinal lymphadenopathy in an 11-year old girl: a case report. Trop Doct, 2015, 45(1):54-56.
[34] Akhavan A, Cannon GM Jr, Sasatomi E, et al. Synchronous unilateral renal cell carcinoma and Kimura disease of the kidney. Urology, 2006, 68(3):673.e21-22.
[35] 陈辉树, 陈华, 张乃鑫, 等. 木村病38年伴发皮肤T细胞淋巴瘤一例. 中华血液学杂志, 2007, 28(4):271-272.
( 本文编辑:李贵存 )
Kimura disease: 1 case report and 81 cases literature review
HU Tan, WU Ji, WU Di, ZHENG Chao, HUANG Rong-rong.
Graduate Division, Dalian Medical University, Dalian, Liaoning, 116044, PRC Corresponding author: WU Ji, Email: bjwuji@sina.com
s】 Objective To explore the epidemiological and pathological characteristics, clinical manifestations, diagnosis and differential diagnosis, clinical treatment and prognosis of Kimura disease by summarizing clinical data of 81 patients with Kimura disease. Methods One case admitted in our department in 2014 was reported. Chinese biological and medical literature databases such as Wanfang, China National Knowledge Infrastructure ( CNKI ) and Vip from January 2000 to December 2014 were searched. Key words were Kimura, Kimura's disease and eosinophilic lymphoid granuloma. Results There were 82 cases ( 63 males and 19 females ) with complete clinical data ( including 1 case in our hospital ). The male female ratio was 3.3:1. The mean age was 39 years, and mean disease course 5.79 years. 78.05% of cases presented with the head and neck subcutaneous mass, and 41.46% were accompanied by regional lymphadenopathy. Simple lymph node enlargement occurred in 21.95% of cases, salivary gland involvement in 21.95%, local symptoms of mass in 40.24%, and related complications in 35.37%. Peripheral blood eosinophilia and elevated serum immunoglobulin E ( IgE ) were found in 85.37% and 42.68% of cases respectively. 28.05% and 18.29% of cases were treated with surgical operation and steroid hormones respectively and 45.12% with combined therapy. Conclusions Kimura disease is a rare chronic inflammatory disorder of unknown etiology, and usually presents as a subcutaneous mass in the head and neck region or the major salivary glands of young to middle-aged Asian men. It is often associated with regional lymphadenopathy. Elevated serum IgE and peripheral blood eosinophilia are also common. Though surgical resection, regional or systemic steroid therapy and local radiation have good effects, recurrence may occur.
Angiolymphoid hyperplasia with eosinophilia; Review literature as topic; Kimura's disease
10.3969/j.issn.2095-252X.2015.09.007
R758.6
116044 辽宁,大连医科大学研究生部 ( 胡袒 );100142 北京,空军总医院骨科 ( 伍骥、吴迪、郑超 ),普外科 ( 黄蓉蓉 )
伍骥,Email: bjwuji@sina.com
2015-05-05 )
