烟草青枯病抗性与分子标记的关联分析
2015-11-27夏岩石李荣华吕永华余义文赵伟才邱妙文郭培国
吴 超,夏岩石,李荣华,吕永华,余义文,赵伟才,邱妙文,郭培国*
1.广州大学生命科学学院,广州市番禺区大学城外环西路230号 510006
2.广东省烟草专卖局(公司),广州市天河区林和东路128号 510610
3.广东省烟草南雄科学研究所,广东省南雄市雄州镇河南工业开发区东厢铺1号 512400
烟草青枯病抗性与分子标记的关联分析
吴 超1,夏岩石1,李荣华1,吕永华2,余义文1,赵伟才3,邱妙文3,郭培国*1
1.广州大学生命科学学院,广州市番禺区大学城外环西路230号 510006
2.广东省烟草专卖局(公司),广州市天河区林和东路128号 510610
3.广东省烟草南雄科学研究所,广东省南雄市雄州镇河南工业开发区东厢铺1号 512400
为了找到与烟草青枯病抗性相关的分子标记、加快抗病品种的选育,以78份烟草种质为自然群体材料,选用20对简单重复序列(Simple Sequence Repeat,SSR)和37对微卫星锚定片段长度多态性(Microsatellite-anchored Fragment Length Polymorphism,MFLP)引物组合对烟草种质材料进行基因分型,并利用等位变异数据进行主成分分析和群体结构分析,同时采用TASSEL3.0软件对烟草青枯病抗性与等位变异进行连锁不平衡关联分析.结果显示:SSR和MFLP标记在78份烟草种质中共检测到252个等位变异,主成分分析和群体结构分析均将烟草种质分为5个组群;关联分析发现6个MFLP标记位点与烟草青枯病抗性显著相关,对青枯病抗性的解释率在8.33%~19.49%之间.这些分子标记可为评价具有青枯病抗性潜力的烟草种质材料提供依据.
烟草青枯病;分子标记;遗传多样性;群体结构;关联分析
烟草青枯病是由青枯雷尔氏菌(Ralstonia solanacearum)引起的一种土传病害[1].近年来随着烟草生产集约化程度的提高,烟草青枯病已成为我国南方烟区烟叶生产上的主要病害之一,并且发病范围有向北方烟区蔓延的趋势[2].目前采用的耕作控制、生物防治、化学防治等措施能够在一定程度上降低青枯病的危害,但均未取得满意的防治效果[3-5],而根据烟草青枯病抗病遗传特性选育出抗病品种则是控制该病害的最为科学有效的途径.有研究表明,烟草青枯病抗性为受多基因控制的数量性状,易受到基因型与环境交互作用(互作)的影响,因此采用传统方法选育青枯病抗病品种的进展较慢[6].分子标记辅助选择技术能减少环境互作的影响,提高筛选效率、加快育种进程,且已成功应用在多种作物上[6-7].基于连锁不平衡的关联分析是近10年来开展作物分子遗传和育种应用研究的一种新方法,以遗传背景差异较大的自然群体为研究对象,经过长期重组保留的基因(或标记)位点间的连锁不平衡为基础,将目标性状表型的多样性与基因(或标记)位点的多态性相结合进行分析,能直接获得与表型相关且具有特定功能的基因(或标记)位点[8];与经典的连锁分析相比,关联分析利用自然群体材料多世代中的重组事件,可检测同一座位的多个等位基因,具有更为广泛的遗传变异和较高的分辨率以及发现复杂性状所涉及到的多基因及其相互关系的潜力[9].目前关联分析已在水稻、小麦和大麦等农作物的一些农艺性状和相关基因研究中得到应用,并取得一定成效[10-12].但在烟草上的研究报道较少,余义文等[13]通过关联分析发现了1个简单重复序列(Simple Sequence Repeat,SSR)和 6个微卫星锚定片段长度多态性(Microsatelliteanchored Fragment Length Polymor-phism,MFLP)标记的多态性位点与烟草特有亚硝胺含量显著相关,任民等[14]找到了24个SSR标记位点与烟草致香物质相关联,但未见有关烟草青枯病抗性关联分析研究的报道.为此,以78份烟草种质材料为自然群体,利用SSR和MFLP标记技术对自然群体各材料进行基因分型,并对自然群体的遗传多样性和群体结构进行分析,同时结合自然群体各材料的抗病数据进行关联分析,旨在发现与烟草青枯病抗性相关的分子标记位点,为烟草抗青枯病的分子标记辅助选择育种提供依据.
1 材料与方法
1.1 材料
供试78份烟草种质材料来源于美国、印尼、毛里求斯、巴西、古巴以及中国(11个省份),包括烤烟、晒烟、白肋烟和雪茄烟4种类型,由广东省烟草南雄科学研究所提供.各烟草种质材料信息见表1.
1.2 烟草青枯病病情调查与分析
供试烟草种质材料于2011年和2013年种植于广东省烟草南雄科学研究所南雄试验田青枯病病圃,每个品种栽种20株,采用随机区组设计.在烟草旺长期采用茎部穿刺接种法人工注射茎秆接种烟草青枯病菌[15],按照标准GB/T 23222─2008调查烟草病害发生情况[16],以成熟期烟草单株为单位,在晴天12:00-16:00调查,然后根据烟草病害等级计算每个烟草种质的青枯病病情指数(DI).
病情指数=∑(各级病株数X该病级值)/(调查总株数X最高级值)X100
依据标准GB/T23224─2008[17]的方法对78份种质进行抗病性鉴定.采用SPSS17.0软件进行表型数据的描述性统计和方差分析,依据环境方差和遗传方差计算广义遗传率(H2).
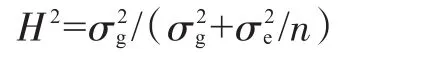
1.3 SSR和MFLP标记全基因组扫描
采集不同种质材料的烟草幼叶,利用李荣华等[18]改进的CTAB方法提取各材料的基因组DNA,采用琼脂糖凝胶电泳和紫外可见分光光度法检测提取的DNA品质和浓度(ng.μL-1).20对SSR选自Bindler等[19]开发的引物,分别位于14条不同的染色体上.利用4个烟草种质(D101、RG17、长脖黄、C151)对8个加尾SSR锚定引物与32种MseI选择性引物组成的256个MFLP引物组合进行扩增分析,从中筛选出条带清晰、多态性好的37个MFLP引物组合用于78份烟草种质分析.荧光SSR和MFLP标记的操作流程及引物名称、序列信息参考文献[20-21].两种标记技术使用的荧光引物M13-F-IRDye 700购自于美国LICOR公司,其他引物均由生工生物工程(上海)股份有限公司合成.
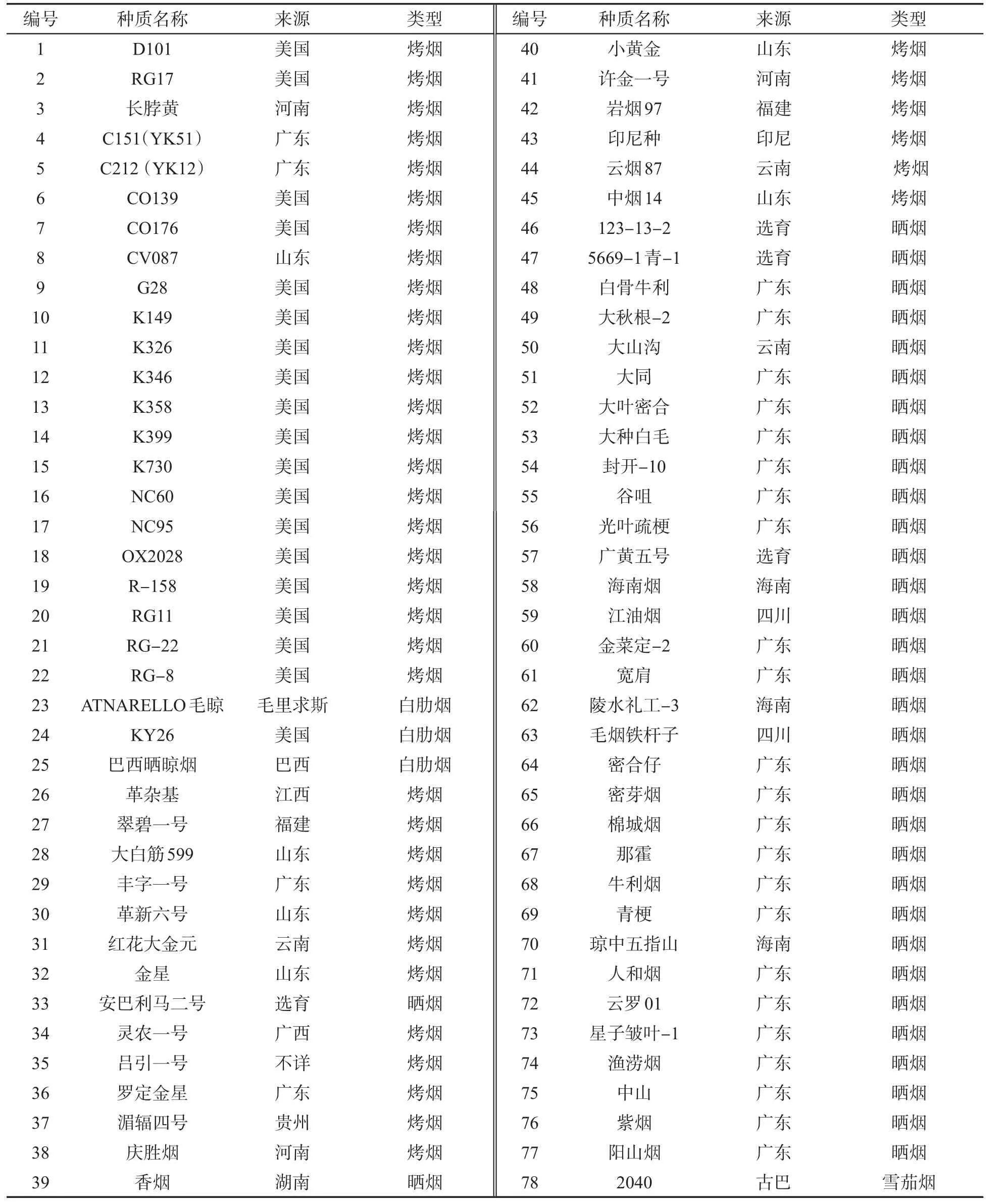
表1 78份烟草种质材料的来源与类型Tab.1 The origins and types of 78 tobacco accessions
1.4 数据处理
对SSR和MFLP电泳图谱进行分析,排除图谱中模糊不清和无法准确标识的条带,每个多态性条带视为一个等位变异位点,在相同碱基大小范围内比较所有样品,有条带记为1,无条带记为0,构建0、1二元数据矩阵.利用PIC-Calc0.6软件计算标记的多态信息量(Polymorphism Information Content,PIC).
利用NTSYS-pc2.1软件的Dcenter和Eigen对供试烟草材料进行主成分分析(Principal Component Analysis,PCA).利用POPGENE Version 1.31软件计算基因多样性与Shannon信息指数.利用Structure 2.3软件对78份烟草材料进行群体结构分析,组群数量(K值)为1~10,将MCMC(Markov Chain Monte Carlo)的不作数迭代(Length of Burn-in Period)设为100 000次,再设置不作数迭代后的MCMC为100 000次,每个K值进行5次计算.依据似然值最大原则选取最合适的K值,绘制群体遗传结构图,并获得关联分析的校正系数(Q值).应用SPAGeDi1.3d软件计算烟草种质间的亲缘系数,并将其中所有负值设为0[22].为获得更准确的关联结果,避免亚群混乱造成的伪关联,以群体结构Q值和亲缘系数值矩阵作为协变量进行群体校正[23],使用TASSEL3.0软件的混合线性模型(Mixed Linear Model,MLM)程序[24],将烟草青枯病病情指数分别与标记变异进行关联分析,找出与烟草青枯病抗性显著关联的SSR标记和MFLP标记位点,并计算标记位点对表型变异的解释率.阈值选择以Plt;0.01为显著水平,Plt;0.001为极显著水平.
2 结果与分析
2.1 烟草青枯病病情的统计与分析
对78份烟草自然群体进行发病株数和发病等级调查,依据每株烟草的发病等级计算各品种的病情指数并进行抗病分级鉴定,其中9个品种在两年的统计中均表现为抗病(R)(0lt;DI≤20),结果见表2.青枯病病情指数的统计分析结果(表3)表明,2011年和2013年烟草青枯病病情指数的变异系数均较大,分别为51.49%和68.99%;两年的病情指数广义遗传率均超过70%,分别为72.29%和73.72%.不同供试烟草种质对青枯病抗性差异较大,说明抗病性变异丰富,适合进行烟草青枯病抗性关联分析,并能够提供丰富的烟草种质资源用于抗青枯病烟草品种的选育.
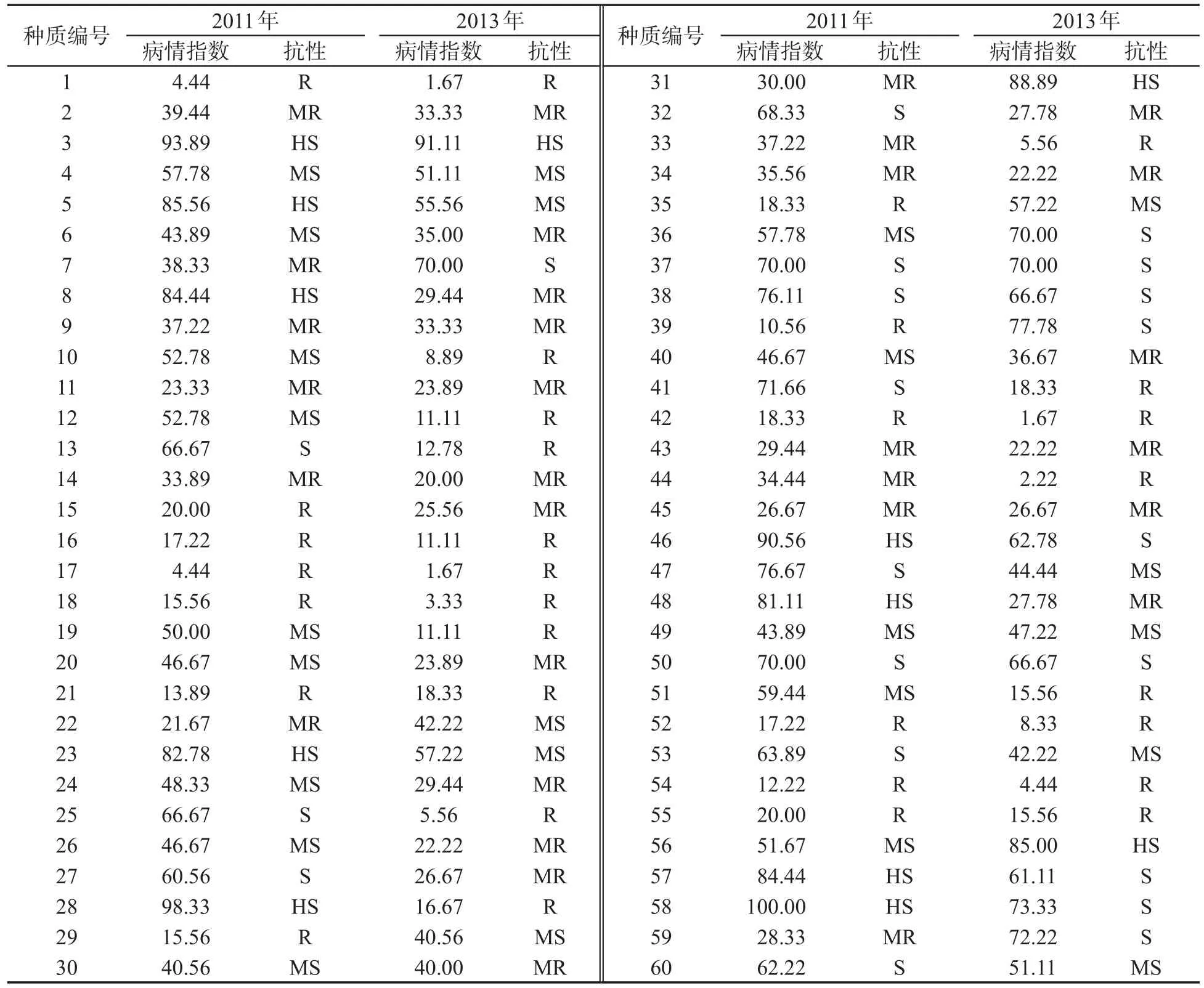
表2 烟草青枯病病情指数与抗性鉴定①Tab.2 Tobacco bacterial wilt disease index and resistance identification
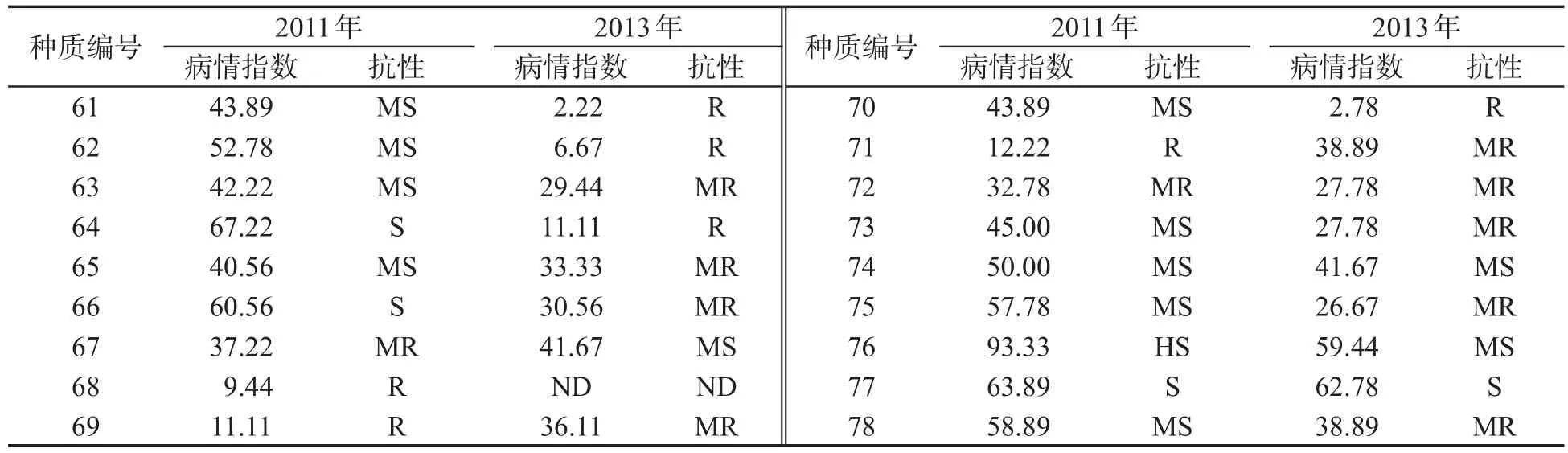
表2(续)
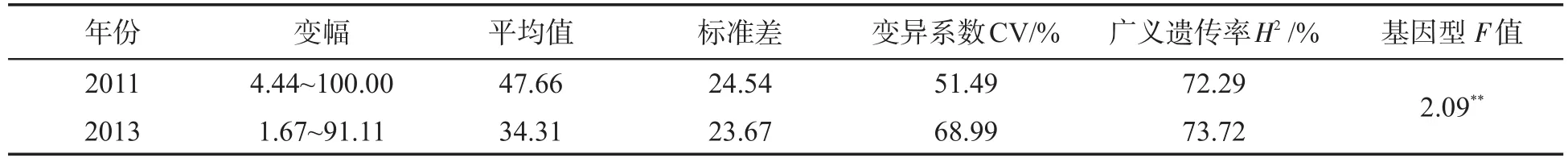
表3 供试烟草青枯病病情指数的统计分析①Tab.3 Statistic data of tobacco bacterial wilt disease index
2.2 扩增产物多态性分析
使用20对SSR引物对78份烟草种质DNA进行扩增,共检测到85个多态性位点,多态性位点数目变化范围2~8个,平均每个SSR标记存在4.25个等位变异.SSR标记的PIC值变幅为0.246 1~0.795 9,平均PIC值为0.542 7.利用37对MFLP引物进行扩增,检测到167个多态性位点,平均每对引物扩增了4.51个多态性位点,PIC变幅为0.313 0~0.913 8,平均PIC值为0.629 8.表明所选SSR引物与MFLP引物具有较高的多态性,用这些引物对供试烟草材料进行扩增可以反映较丰富的多态性信息.具体每个SSR引物与MFLP引物扩增的多态位点数和PIC,见表4.利用两种分子标记共检测到252个等位变异位点.
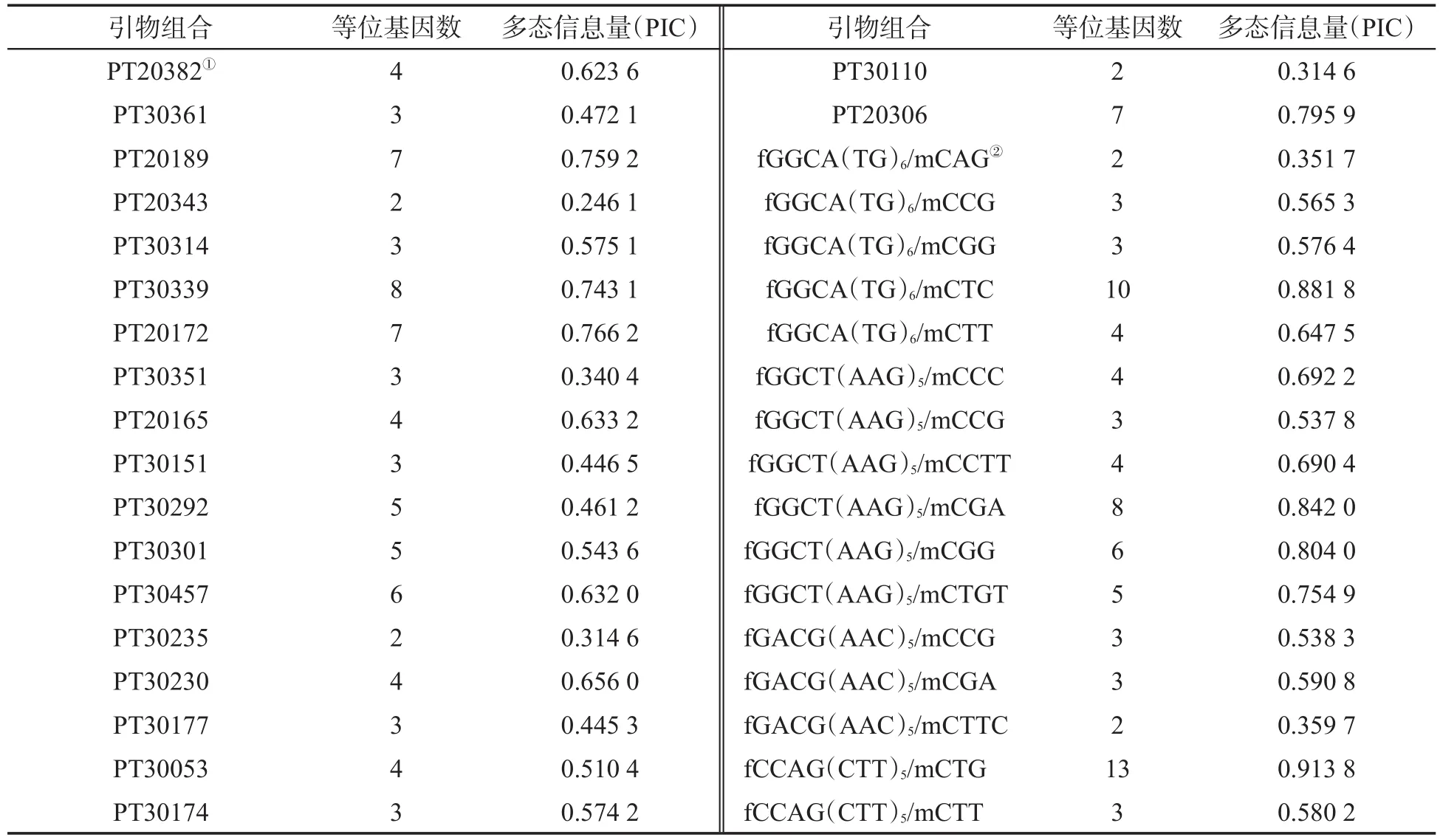
表4SSR标记与MFLP标记的等位变异分析Tab.4 Allelic variation analysis of SSR markers and MFLP markers
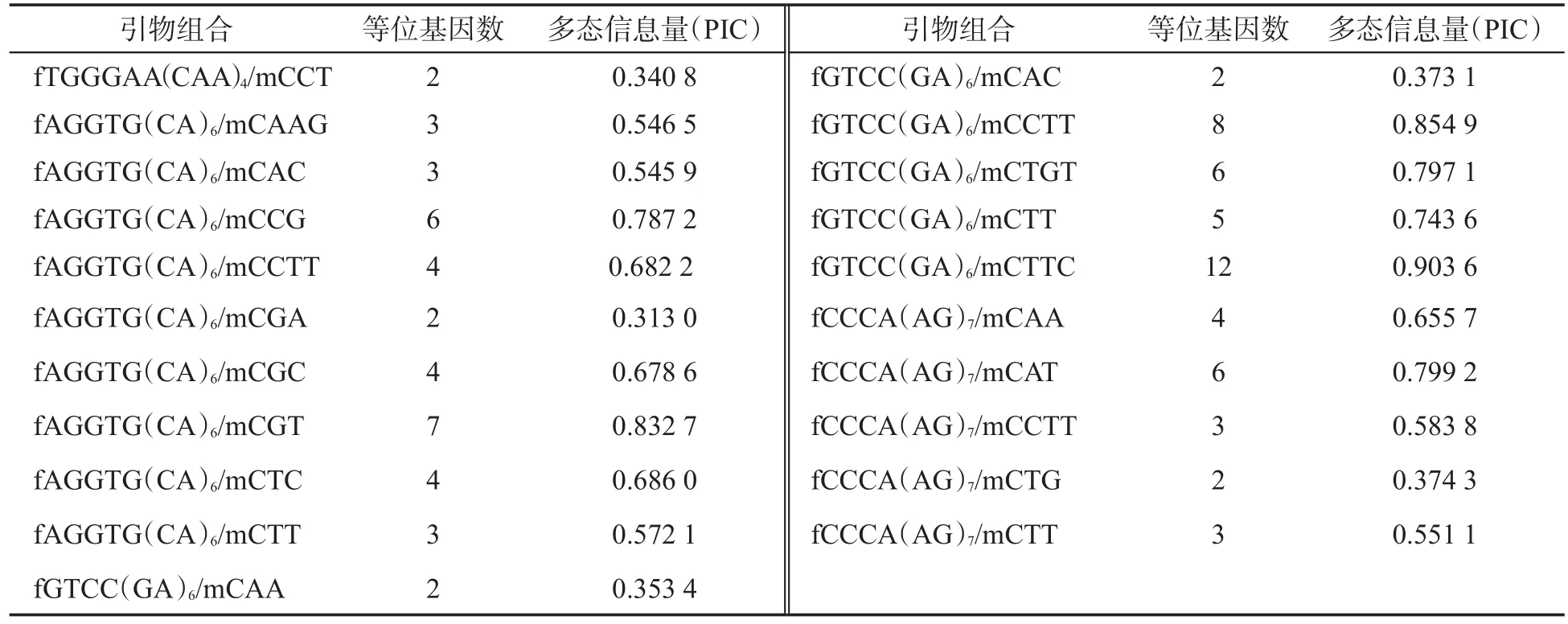
表4(续)
2.3 烟草种质的主成分分析
对78份供试烟草种质材料进行基于0、1数据矩阵的PCA分析,第1主成分能够解释的遗传变异为20.38%,第2和第3主成分解释的遗传变异分别为11.45%和10.87%,合计为42.70%.以78份材料的三维主成分数据绘制三维分布图(图1).根据位置近则亲缘关系近,位置远则亲缘关系远的原则[25],78份烟草种质可分为5个类群.类群a包含5份烟草种质,其中我国晒烟和烤烟品种各2份,另1份为美国烤烟品种NC60;类群b共有23份烟草种质,包括除NC60以外的全部17份供试美国烤烟品种和6份我国的地方烤烟品种;类群c包括24份烟草种质,含有供试的3份不同国家的白肋烟品种、1份古巴雪茄烟品种、8份晒烟品种以及12份烤烟品种;类群d含有18份烟草种质,除云烟87以外均为晒烟品种,其中14份为广东晒烟品种;类群e包含8份烟草种质,7份广东晒烟和1份海南晒烟品种.从PCA分析结果来看,供试的23份广东晒烟中有21份被归类到d和e类群,占供试广东晒烟种质的91.3%;广东晒烟与美国烤烟分别归属到了不同的类群中,二者间的遗传距离较远.
2.4 种质群体结构与基因多样性分析
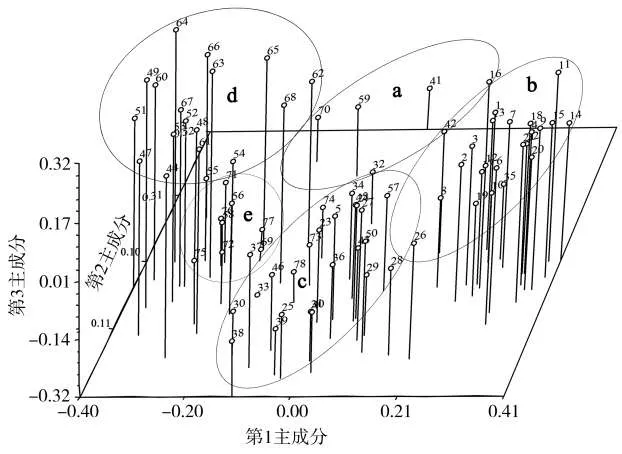
图1 78份烟草种质材料的三维主成分分析Fig.1 Three-dimension principal component analysis of 78 tobacco accessions
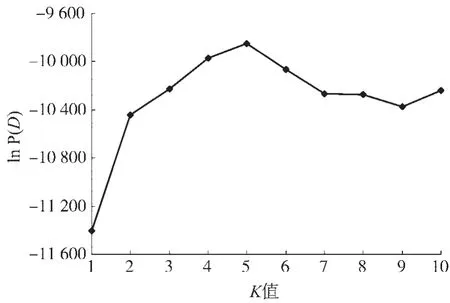
图2K值与ln P(D)值分布图Fig.2 Distribution chart ofKand ln P(D)
利用Structure 2.3软件对78份烟草种质材料进行群体结构分析.K值取1~10,以K值为横坐标,相应ln P(D)值为纵坐标做图.结果(图2)显示:78份烟草种质的等位变异频率特征类型数K=5(即服从Hardy-Weinberger平衡的组群数目为5)时其ln P(D)值最大,表明当K=5时其模型后验概率最大.因此78份烟草种质材料可分为5个组群,群体结构如图3所示.
对各烟草种质材料在不同组群的Q值进行分析,其中66个种质材料在所属组群的Q值大于0.5,占全部供试材料的84.62%,这些种质遗传组分相对单一,可归入5个组群中某一个,另外12个种质在组群中的Q值均小于0.5,列为混合群体,其组群归属特征不明显(表5).
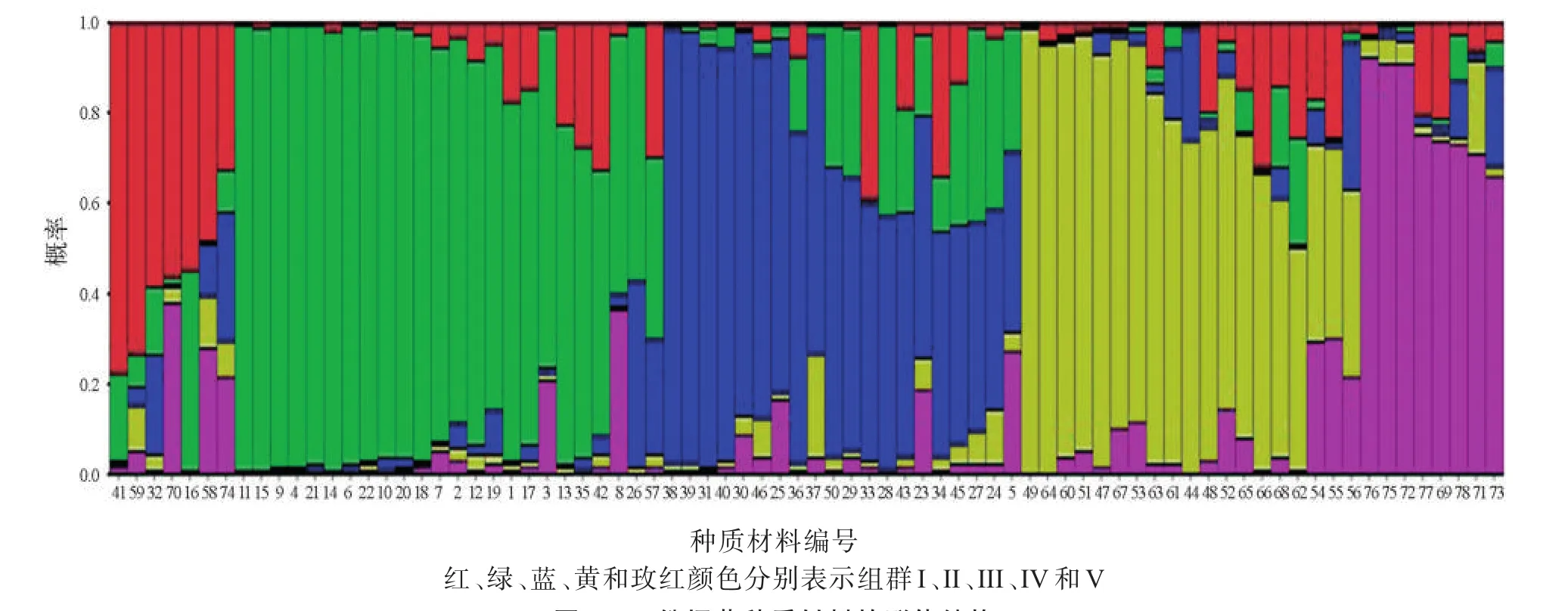
图3 78份烟草种质材料的群体结构Fig.3 Population structure of 78 tobacco accessions
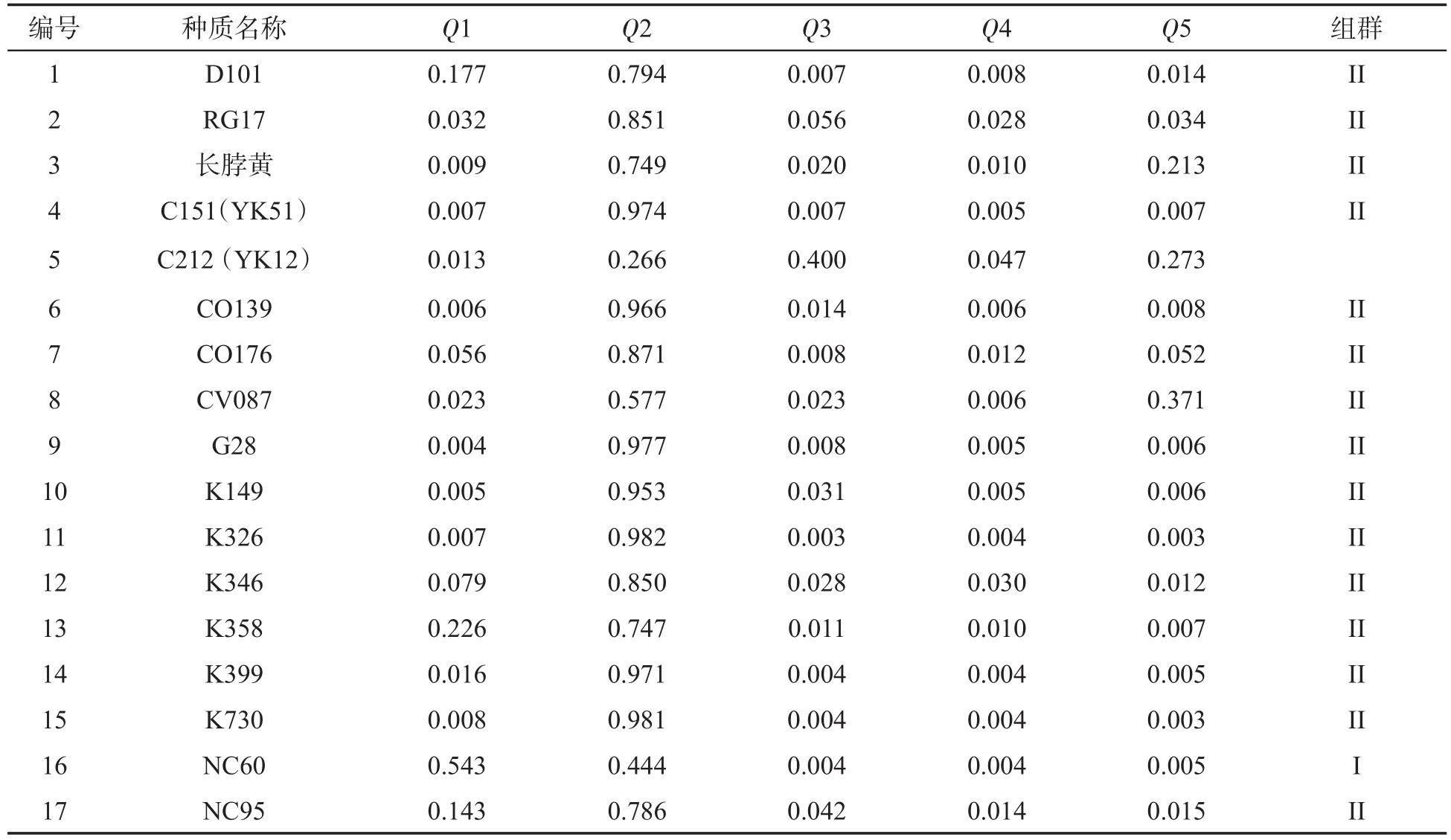
表5 78份烟草种质的群体结构Tab.5 Population structure of 78 tobacco accessions
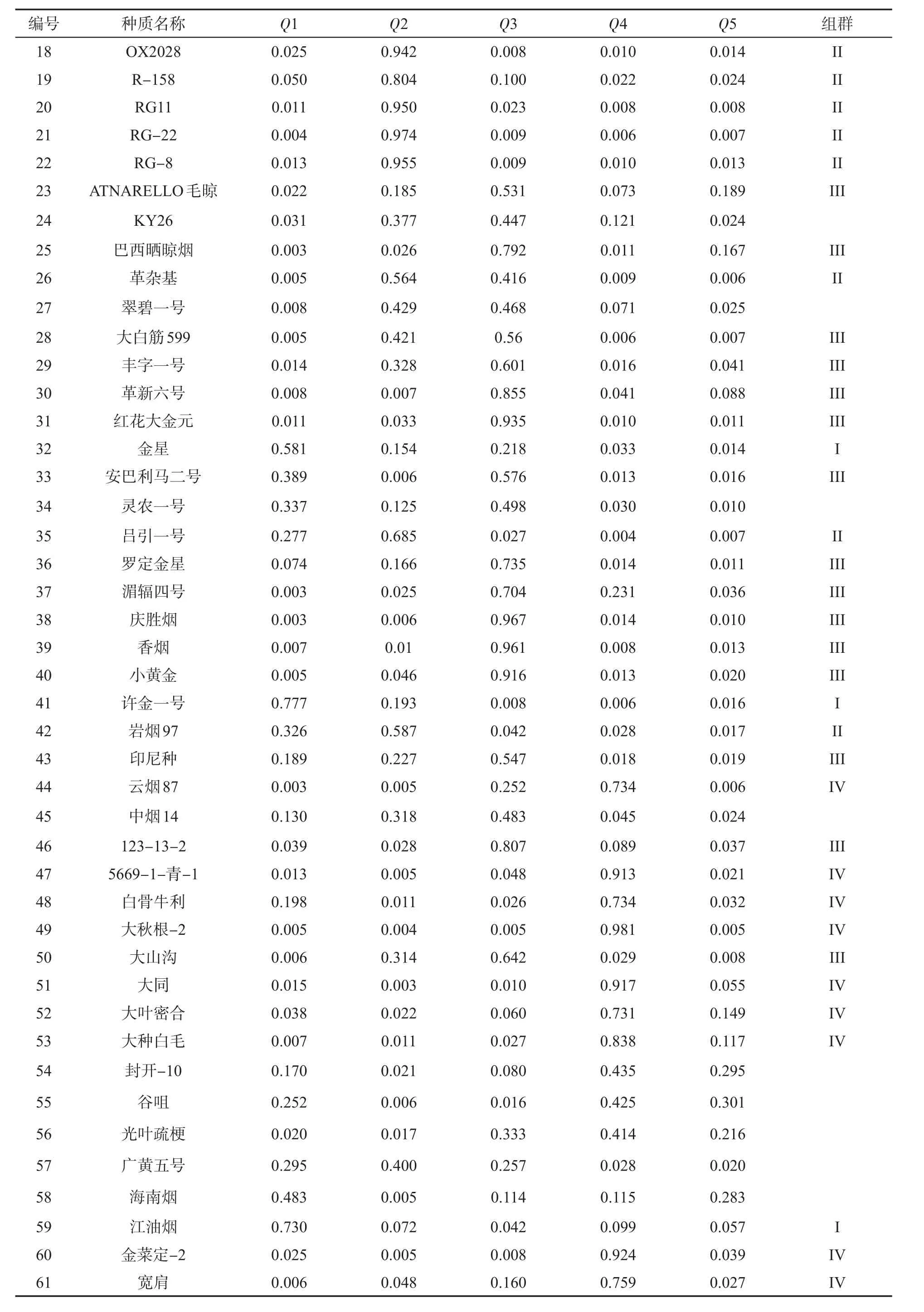
表5(续)
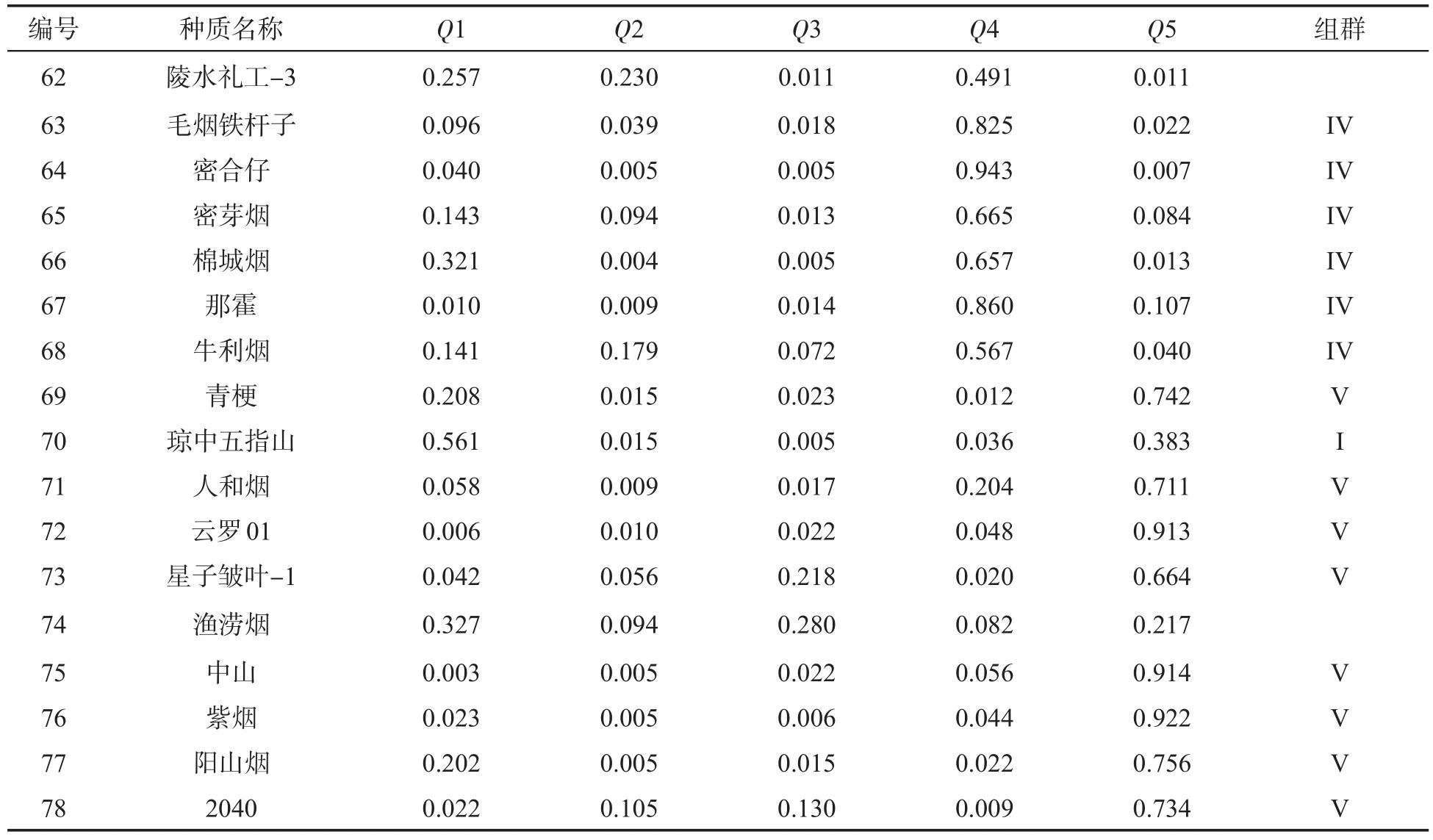
表5(续)
对66份归入指定组群的烟草种质材料进行分析表明,在5个组群中,组群I包括5个烟草种质,其中有3个烤烟和2个晒烟.组群II包括23个烟草种质,全部为烤烟,其中包含了除NC60以外的全部17个供试美国烤烟品种与6个中国烤烟品种.组群III包括15个烟草种质,其中包括2个白肋烟、9个烤烟、4个晒烟.组群IV包括15个烟草种质,其中包括14个晒烟和1个云南烤烟(云烟87),该组群14个晒烟中有12个为广东晒烟品种.组群V包括8个烟草种质,其中7个为广东晒烟.组群IV和V中绝大多数为广东晒烟.PCA分析中类群c的24个烟草种质中除组群III包含的15份种质材料外,另有5份在组群III的Q值最大,4份在组群III的Q值较大;类群d的18个种质中包括了组群IV的15份种质材料和3个在组群IV的Q值最大的种质.群体结构分析结果与PCA分析结果基本一致.
种质材料间的亲缘系数计算结果显示:两种质间亲缘系数位于0~0.05之间的组合占总组合的73.96%,其中54.11%亲缘系数为0,所有亲缘系数均小于0.5.表明大多数供试种质材料间亲缘关系较远或无亲缘关系.对利用群体结构分析产生的5个组群进行基因多样性和Shannon指数分析,基因多样性大小和Shannon指数大小基本一致,5个组群的基因多样性由大到小依次为组群IIIgt;组群Vgt;组群IVgt;组群Igt;组群II,Shannon指数大小依次为组群IIIgt;组群Vgt;组群IVgt;组群IIgt;组群I.对各组群的基因多样性和Shannon指数分别进行了差异显著性分析,结果见表6.从表6中可见,组群IV和组群V间基因多样性与Shannon指数均无显著差异,遗传距离相对较近,而在PCA图上距离较远的群体差异显著.
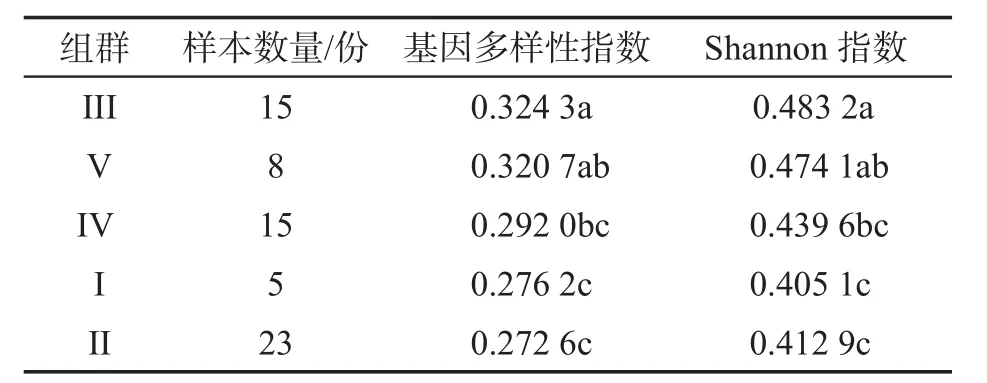
表6 组群的遗传多样性①Tab.6 Summary statistics of genetic diversity of groups
2.5 烟草青枯病抗性与分子标记的关联分析
将78份烟草群体结构分析中K=5时对应的Q值以及计算的亲缘系数值作为关联分析的协变量,将252个标记位点与供试烟草材料的青枯病病情指数进行关联分析,结果见表7.发现6个标记位点与烟草青枯病抗性存在显著关联,其中MFLP标记位点M142与2011年和2013年病情指数的统计数据均显著相关,对表型变异的解释率分别为10.42%和9.04%.标记M184和M221与2013年青枯病病情指数显著相关,M146、M206和M237与2011年青枯病病情指数显著相关.各标记位点对青枯病抗性表型变异的解释率在8.33%~19.49%之间,平均解释率为11.08%.
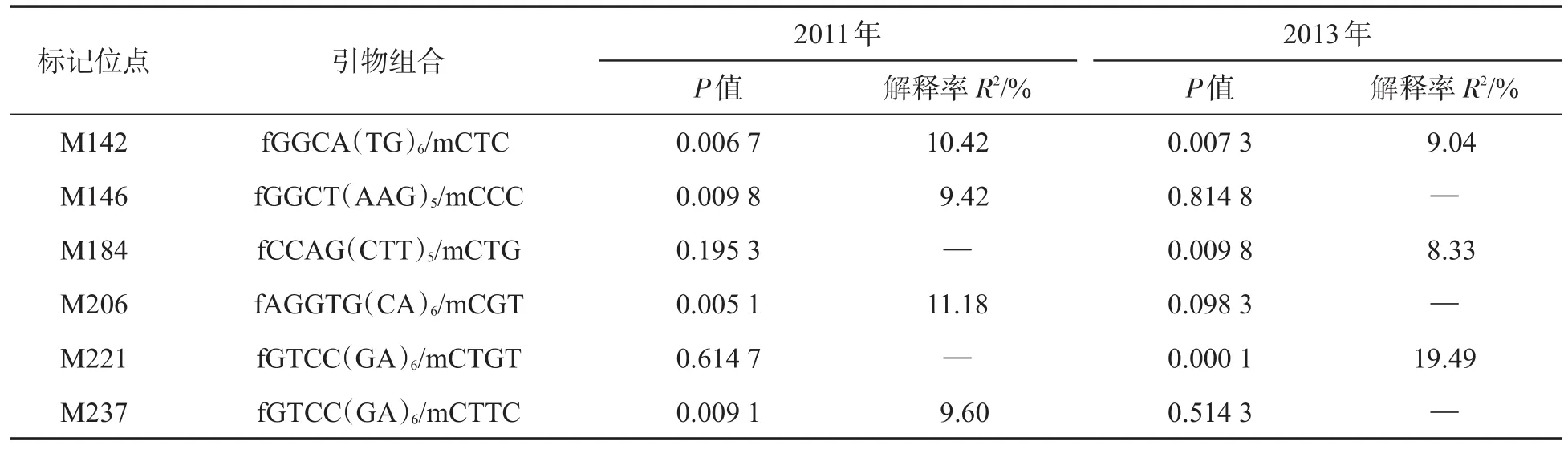
表7 与烟草青枯病病情指数关联的标记位点及对表型变异的解释率Tab.7 Marker loci associated with tobacco disease index of bacterial wilt and their explained portion of phenotypic variation
3 结论与讨论
群体结构的存在会通过对等位变异位点(LD)的影响而影响关联分析的准确性.在不考虑试验材料群体结构的情况下,组群的混合使整个自然群体所估计的LD强度增强,有可能造成表型性状与多态位点的关联性并不是由功能性等位基因引起,从而造成假关联[26].纳入结构分析后的Q值可矫正关联分析的偏差,避免了组群混合造成的伪关联[27].在本试验中利用252个分子标记等位变异数据,对供试烟草种质材料进行的群体结构分析在K值为5时ln P(D)值出现拐点,从而将78份烟草分为5个组群.绝大多数烤烟与晒烟品种被归到不同组群,表明烤烟与晒烟之间具有一定的遗传差异.从美国烤烟与部分中国烤烟分在同一组群,以及中国不同省份烤烟分在同一组群这一结果可以看出,不同地区烤烟的遗传多样性差异并不大.广东晒烟分布在两个组群,与其他地区晒烟组群不同,表明广东晒烟与其他地区晒烟间的遗传差异较大,该结果与何其芳等[20-21]的研究结论基本一致;亦与早期报道的我国烟草材料不同烟草类型间遗传差异较大,烤烟遗传多样性水平较低,晒烟遗传变异较丰富的研究结果基本一致[28-29].另外,种质材料的PCA分析与群体结构分析结果基本一致,两种方法的结合以及相互验证,提高了试验结果的可靠性;同时PCA分析结果进一步证明了K值为5时群体结构划分的合理性,用该条件下的各个体Q值作为关联分析的一个协变量是合理的.
烟草青枯病抗性属于典型的数量性状遗传,由多基因共同控制,并受环境因素影响较大,准确统计烟草的抗青枯病表型数据对研究抗病标记至关重要[30].本研究中以种植于南雄烟科所试验田青枯病病圃的78份亲缘关系较远的烟草种质为研究材料,采用国标中的病情统计和鉴定方法进行分析,避免了标准不统一造成的误差,保证了表型数据的可靠性.对病情指数的分析结果表明:2011年和2013年病指的变异系数均大于50%,种质间遗传差异大,适用于关联分析.对群体的基因分型所得0、1矩阵进行了Kinship分析,再次验证了供试种质间亲缘关系较远.正确的自然群体结构对于有效控制假阳性、提高关联分析可靠性至关重要.以群体结构Q值和亲缘系数值作为协变量,采用MLM_Q+亲缘系数值模型进行关联分析,发现了M142、M146和M184等6个与烟草抗病显著关联的MFLP标记位点,其中标记M142与两年的病指均显著关联.有研究表明MLM_Q+亲缘系数值模型为烟草群体关联分析的最优模型之一,能够有效控制假阳性的产生[23],因此研究结果可靠性较高.
筛选与目的基因相关的分子标记是分子标记辅助选择(MAS)育种的前提,连锁分析和基于连锁不平衡的关联分析是现今解析复杂性状遗传、发现与目标基因紧密连锁或共分离关系分子标记、进而开发出分子标记并用于MAS育种的主要方法[26].已有一些研究报道了利用两个烟草亲本的杂交后代作材料,对青枯病抗病性进行了连锁分析并取得了一些成绩,发现一些与青枯病抗性密切相关的数量性状位点(QTLs)[6,31-33].由于连锁分析在构建分离群体时受杂交和自交次数的限制,发生的重组次数较少,另外连锁分析仅涉及同一座位的两个等位基因;而青枯病抗性是由多基因控制的复杂性状,每一个基因可能对抗病性起着微效作用,且有些基因的外显力低,受环境的影响较大,连锁分析方法难以完全解析这一复杂的关系[26].因此,现阶段通过连锁分析定位青枯病抗病QTLs的结果与离分子标记辅助选择育种的要求有一定的距离[34].而基于连锁不平衡的关联分析对发现复杂性状所涉及的多基因及相互关系具有较大的潜力,这种方法选用遗传背景差异大的自然群体为研究对象,检测多世代重组事件中同一座位的多个等位基因,并对等位变异与表型进行关联分析,能较准确地发现等位变异与性状的关系[25],是现今解析植物数量性状遗传的主要方法之一.本研究中利用SSR和MFLP标记对烟草种质材料进行了烟草青枯病抗性关联分析,发现了与青枯病抗性相关的6个MFLP分子标记位点,这一结果可为烟草抗青枯病的分子标记辅助选择育种提供理论依据.
[1]Tans-Kersten J,Guan Y,Allen C.Ralstonia solanacearum pectin methylesterase is required for growth on methylated pectin but not for bacterial wilt virulence[J].Applied and Environmental Microbiology,1998,64(12):4918-4923.
[2] 孔凡玉.烟草青枯病的综合防治[J].烟草科技,2003(4):42-43,48.
[3] 周训军,王静,杨玉文,等.烟草青枯病研究进展[J].微生物学通报,2012,39(10):1479-1486.
[4] 刘先良,习向银,申鸿,等.接种丛枝菌根真菌对烟草青枯病抗性的影响[J].烟草科技,2014(5):94-98.
[5] 万川,蒋珍茂,赵秀兰,等.深翻和施用改良剂对烟草青枯病发生的影响[J].烟草科技,2015,48(2):11-15,26.
[6]QIAN Yiliang,WANGXinsheng,WANGDazhou,et al.The detection of QTLs constrolling bacterial wilt resistance in tobacco(N.tabacum L.)[J].Euphytica,2013,192(2):259-266.
[7] XIAYanshi,LIRonghua,NING Zhengxiang,et al.Single nucleotide polymorphisms in HSP17.8 and their association with agronomic traits in barley[J].PLoS ONE,2013,8(2):e56816.
[8] March R E.Gene mapping by linkage and association analysis[J].Molecular Biotechnology,1999,13(2):113-122.
[9] Yu J,Buckler E S.Genetic association mapping and genome organization of maize[J].Current Opinion in Biotechnology,2006,17(2):155-160.
[10]Iwata H,EbanaK,Uga Y,etal.Genome-wide association study of grain shape variation among Oryza sativa L.germplasms based on elliptic Fourier analysis[J].Molecular Breeding,2010,25(2):203-215.
[11]Neumann K,Kobiljski B,Denčić S,et al.Genome-wide associationmapping:A casestudyinbreadwheat(Triticum Aestivum L.)[J].Molecular Breeding,2011,27(1):37-58.
[12]XIA Yanshi,NING Zhengxiang,BAI Guihua,et al.Allelic variations of a light harvesting chlorophyll a/b-binding protein gene(Lhcb1)associated with agronomic traits in barley[J].PLoS ONE,2012,7(5):e37573.
[13]余义文,夏岩石,李荣华,等.烟草种质材料TSNA含量的关联分析[J].中国烟草学报,2014,20(3):48-55.
[14]任民,张长静,蒋彩虹,等.基于高密度SSR连锁群的烟草致香物质关联分析[J].中国烟草学报,2014,20(4):88-93.
[15]申莉莉.烟草突变体筛选与鉴定方法篇:2.烟草抗主要病虫害突变体的筛选与鉴定[J].中国烟草科学,2012,33(2):102-104.
[16]GB/T 23222-2008 烟草病虫害分级及调查方法[S].
[17]GB/T 23224-2008 烟草品种抗病性鉴定[S].
[18]李荣华,夏岩石,刘顺枝,等.改进的CTAB提取植物DNA方法[J].实验室研究与探索,2009,28(9):14-16.
[19]Bindler G,van der Hoeven R,Gunduz I,et al.A microsatellite marker based linkage map of tobacco[J].Theoreticaland AppliedGenetics,2007,114(2):341-349.
[20]何其芳,李荣华,郭培国,等.利用荧光MFLP标记技术分析烟草种质的遗传多样性[J].中国烟草科学,2012,33(1):12-18.
[21]何其芳,李荣华,郭培国,等.利用SSR荧光标记技术分析烟草种质的遗传多样性[J].中国农学通报,2012,28(10):95-102.
[22]YuJ,PressoirG,Briggs W H,etal.A unified mixed-modelmethod forassociation mapping that accounts for multiple levels of relatedness[J].Nature Genetics,2006,38(2):203-208.
[23]张吉顺,王仁刚,杨元春,等.国内外烤烟品种农艺性状的遗传多样性及SRAP标记的关联分析[J].作物学报,2012,38(6):1029-1041.
[24]Bradbury P J,Zhang Z,Kroon D E,et al.TASSEL:software for association mapping of complex traits in diverse samples[J].Bioinformatics,2007,23(19):2633-2635.
[25]陈斐,魏臻武,李伟民,等.基于SSR标记的苜蓿种质资源遗传多样性与群体结构分析[J].草地学报,2013,24(4):759-768.
[26]Mackay I,Powell W. Methods for linkage disequilibrium mapping in crops[J].Trends in Plant Science,2007,12(2):57-63.
[27]Flint-Garcia S A,Thuillet A C,Yu J,et al.Maize association population:a high-resolution platform for quantitative trait locus dissection[J].Plant Journal,2005,44(6):1054-1064.
[28]杨友才,周清明,尹晗琪.利用RAPD和AFLP标记分析烟草种质资源的遗传多样性[J].农业生物技术学报,2006,14(4):585-593.
[29]肖炳光,杨本超.利用ISSR标记分析烟草种质的遗传多样性[J].中国农业科学,2007,40(10):2153-2161.
[30]吴超,夏岩石,吕永华,等.烟草青枯病抗性分子标记研究进展[J].分子植物育种,2015,13(4):937-945.
[31]Nishi T,Tajima T,Noguchi S,et al.Identification of DNA markersoftobacco linked to bacterialwilt resistance[J].Theoretical and Applied Genetics,2003,106(4):765-770.
[32]杨友才,周清明,朱列书.烟草青枯病抗性基因的遗传分析及RAPD标记[J].中国烟草学报,2006,12(2):38-42.
[33]范江,刘勇,童治军,等.烤烟品种'Oxford207'青枯病抗性的遗传分析与分子标记初选[J].中国农学通报,2013,29(34):50-55.
[34]刘勇,范江,李永平.烟草抗青枯病育种研究进展[J].中国烟草学报,2012,18(6):93-98.
责任编辑 董志坚
Association Analysis of Tobacco Bacterial Wilt Resistance with Molecular Markers
WU Chao1,XIA Yanshi1,LI Ronghua1,LÜ Yonghua2,YU Yiwen1,ZHA O Weicai3,QIU Miaowen3,and GUO Peiguo*1
1.School of Life Sciences,Guangzhou University,Guangzhou 510006,China
2.Guangdong Tobacco Monopoly Administration,Guangzhou 510610,China
3.Nanxiong Tobacco Science Research Institute of Guangdong Province,Nanxiong 512400,Guangdong,China
In order to find out the molecular markers related to tobacco bacterial wilt(TBW)resistance and speed up the breeding process of cultivars resistant to TBW,78 tobacco accessions were selected as natural population,genotyping was performed with 20 pairs of simple sequence repeat(SSR)primers and 37 pairs of microsatellite-anchored fragment length polymorphism(MFLP)primers.Principal component analysis and population structure analysis were conducted with allelic variation data,and the linkage disequilibrium association analysis between TBW resistance and allelic variants was carried out by TASSEL3.0 software.The results showed that 252 allelic variants were detected by SSR and MFLP in 78 tobacco accessions,these accessions were classified into 5 groups by both principal component analysis and population structure analysis.Association analysis indicated that 6 MFLP loci significantly associated with TBW resistance,which explained 8.33%-19.49%of the phenotypic variation of TBW resistance.These molecular markers can be used as reference indexes for evaluating the tobacco accessions with potential TBW resistance.
Tobacco bacterial wilt;Molecular marker;Genetic diversity;Population structure;Association analysis
S432.41
A
1002-0861(2015)10-0001-12
10.16135/j.issn1002-0861.20151001
2014-12-12
2015-04-30
广东省烟草专卖局(公司)科技计划项目quot;连锁和连锁不平衡联合作图技术在烟草青枯病抗性育种上的应用quot;(201201);quot;烟草抗青枯病育种及分子标记辅助选择研究quot;(201403).
吴超(1987-),在读硕士研究生,研究方向:烟草分子生物学.E-mail:wuchao02207101@foxmail.com;*
郭培国,E-mail:guopg@yahoo.com;guopg@gzhu.edu.cn
吴超,夏岩石,李荣华,等.烟草青枯病抗性与分子标记的关联分析[J].烟草科技,2015,48(10):1-12.WU Chao,XIA Yanshi,LI Ronghua,et al.Association analysis of tobacco bacterial wilt resistance with molecular markers[J].Tobacco Scienceamp;Technology,2015,48(10):1-12.
