抗痛风药物的临床研究进展
2015-09-26张利郭晔堃钟静芬
张利 郭晔堃 钟静芬
(上海医药工业研究院 上海 200437)
抗痛风药物的临床研究进展
张利 郭晔堃 钟静芬*
(上海医药工业研究院 上海 200437)
痛风是一种常见的风湿性疾病,目前,急性痛风常用非甾体抗炎药、秋水仙碱,但它们对伴有并发症的痛风患者无效;而在痛风发作的其他时期则可使用降尿酸药物。近年来黄嘌呤氧化酶(XOD)抑制剂和嘌呤核苷磷酸化酶(PNP)抑制剂等可抑制尿酸合成的药物成为这个领域的研究热点。本文主要介绍了白细胞介素1抑制剂(IL-1β抑制剂)、lesinurad、XOD抑制剂、PNP抑制剂的作用机制,临床效果和不良反应。
XOD抑制剂 IL-1β抑制剂 lesinurad PNP抑制剂
痛风是嘌呤类物质代谢紊乱、血尿酸浓度持续增高导致尿酸盐结晶沉积软组织所致的一组代谢类疾病。痛风发病率逐年升高,但有效且不良反应少的药物很少。痛风的治疗成为全世界医疗界的难题。
痛风治疗的基本目标是降低并维持血清尿酸水平(<6.8 mg/dl),使现有的尿酸盐结晶溶解并不再进一步形成结晶,减少甚至消除痛风发作[1]。
临床上将痛风分为四个时期:①无症状的高尿酸血症;②急性痛风关节炎:病人会在受累关节部位出现剧痛;③症状消失的发作间期;④慢性痛风关节炎:尿酸结晶沉积在软骨、滑液膜及软组织中,形成痛风石。急性期痛风常用抗炎的药物,其他时期一般用促进尿酸排泄或抑制尿酸合成等降低高尿酸血症的药物来预防痛风的急性发作[2]。
1 治疗急性期痛风药物
秋水仙碱和非甾体抗炎药(non-steroidal antiinflammatory drugs, NSAIDs)是治疗急性期痛风的常用药物。秋水仙碱是治疗急性痛风性关节炎的首选药物,其可以抑制关节发炎部位的白细胞聚集,使白细胞吞噬尿酸的作用减弱,减轻局部白细胞破坏引起的炎症反应,而达到迅速消炎的目的。但临床上常会引起胃肠道反应和肝、肾功能损害。非甾体抗炎药抑制环氧化酶的活性,阻断前列腺素(PG)的合成,发挥解热,抗炎,镇痛功效。治疗急性期痛风的NSAIDs主要有对乙酰氨基酚,双氯芬酸钠,吲哚美辛等。
尿酸盐结晶诱导巨细胞激活天冬氨酸蛋白水解酶,催化白细胞介素-1β前体(Pro-IL-1β)转换成IL-1β。IL-1β公认为是引起痛风性关节炎的一个关键的细胞因子,对于痛风的治疗有重要的影响。任何直接或者间接阻断IL-1β与受体结合的药物都可以阻断IL-1β的作用。
目前对白细胞介素-1β抑制剂(IL-1β抑制剂)治疗急性期痛风的研究获得可喜的结果。有卡那单抗冻干粉针剂(canakinumab,Ilaris),利纳西普(rilonacept,Arcalyst)冻干粉针剂和阿那白滞素(anakinra,Kineret)。这三个药物被FDA批准用来治疗周期性发热综合征(cryopyrin-associated periodic syndromes,CAPS),而阿那白滞素还被FDA批准用来治疗风湿性关节炎。
卡那单抗是一个完全人源化的抗IL-1β的单克隆抗体,可结合IL-1β而阻断其与受体结合。半衰期是21~28 d,每8周给药一次。最近,Ⅲ期临床[3]显示其治疗急性痛风的有效性。研究中150 mg的卡那单抗与40 mg的曲安奈德作比较,平均72 h视觉模拟评分疼痛分(visual analogue scale pain score )为25.0 mm vs 35.7 mm,差异为-10.7 mm,卡那单抗明显低(P<0.000 1)。受试者中不良事件卡那单抗为66%,曲安奈德为53%。但这项研究存在一些争议,因为在美国,单剂量注射曲安奈德不是治疗急性痛风的标准治疗方法。
利纳西普在降尿酸初期,可预防痛风的急性发作。在评价利纳西普有效性和安全性的Ⅱ期临床试验中[4],采用随机对照双盲法。利纳西普组(n=41)与对照组(n=42)基线资料无差异。试验周期为16周。试验组每周予皮下注射利纳西普一次(负荷量320 mg,以后每周160 mg),对照组注射安慰剂。治疗初始均给别嘌醇300 mg/d,至血清尿酸浓度低于6 mg/dl。在第12周(初始有效终点),利纳西普组痛风急性发作的患者比例显著低于对照组(14.6% vs 78.57%,P=0.001)。在初始治疗后4周利纳西普组的痛风急性发作率即低于对照组(P=0.007);12周以后利纳西普组痛风急性发生率亦低于对照组(14.6% vs 45.2%,P=0.004)。16周后均停药,之后随访6周未见痛风急性发作反弹。两组间不良反应相当,无死亡及严重感染不良反应报道。最常见的不良反应是感染(利纳西普组14.6%,对照组26.2%)及骨骼肌异常(利纳西普组14.6%,对照组21.4%)。利纳西普组的12周评估完成率要高于对照组(98% vs 79%,P=0.015)。
阿那白滞素是短效的重组IL-1β受体拮抗剂,皮下注射100 mg用于风湿性关节炎患者,半衰期是4~6 h。在一项开放性试验研究中[5],10名使用降尿酸药物后尿酸复发性增高患者或痛风患者为受试者,抗炎药物对他们不起作用或者有严重不良反应。阿那白滞的临床疗效是通过用药前后肿胀和疼痛关节的数目以及患者对疼痛减轻的客观评价来评估的。使用阿那白滞素后的第三天,10名受试者的疼痛平均减少了79%,90%患者体检受累关节完全康复。尽管在对照试验中阿那白滞素还没有广泛用于痛风患者,但是已经用作标签外的痛风治疗药物。
2 治疗高尿酸血症药物
2.1 促进尿酸排泄的药物
这类药物通过抑制近端肾小管对尿酸的重吸收而起作用。大多数痛风患者尿酸排泄少,因此,肾功能正常或轻度异常、无尿路结石及尿酸盐肾病的患者可选用排尿酸药。苯溴马隆服药24 h后尿酸水平为服药前的66%。丙磺舒主要在痛风发作间期和慢性期使用以控制高尿酸血症。
阿斯利康公司在研的lesinurad是一种选择性尿酸再吸收抑制剂,通过促进尿酸从体内排泄来治疗痛风,是新型痛风治疗药物中的主要药物,适用于单独使用别嘌醇后尿酸水平没有降到理想值、对别嘌呤不耐受的患者。
尿酸盐重吸收转运子1(urate reuptake transporter, URAT1)被认为是存在于肾脏中用于转运尿酸盐的主要蛋白,能将尿酸从管腔转运到近曲小管上皮细胞并转化为单羧酸盐。lesinurad可抑制URAT1,通过使尿酸排泄正常化及降低血清尿酸水平来缓解疼痛症状。
2011年5月的安全性、有效性分析的双盲临床Ⅱb期试验[6],有208名患有高尿酸血症和痛风的受试者参加。意向治疗(Intent-to treat)分析数据显示,别嘌醇+200 mg lesinurad联合用药组与别嘌醇+400 mg lesinurad联合用药组的反应应答率分别为63%和74%。而别嘌醇+安慰剂组的反应应答率仅为25%。lesinurad药物虽然仅仅处于Ⅲ期试验阶段,但是该药物临床药效明显,阿斯利康制药公司表示其将会成为新一代治疗痛风病症的畅销药物。
2.2 抑制尿酸合成的药物
黄嘌呤氧化酶(XOD)催化黄嘌呤和次黄嘌呤生成尿酸的过程是痛风和高尿酸血症药物研究的关键靶点。
别嘌醇是作用于黄嘌呤氧化酶中黄嘌呤结合位点的抑制剂,是酶的底物类似物[7]。但是不良反应多,有肝脏、骨髓毒性。
日本帝人公司的非布司他(febuxostat)是非嘌呤类的黄嘌呤氧化酶抑制剂,通过占据酶的疏水空腔从而阻止与黄嘌呤的结合[8]。2009年FDA批准上市,2013年我国CFDA批准上市。非布司他不同于别嘌醇,作用靶标明确,作用机制清晰,给药时无需考虑食物和抗酸剂的影响。轻、中度肝功能不全的患者无需调整剂量。
托匹司他(topiroxostat)是继非布司他之后的又一非嘌呤类的黄嘌呤氧化酶抑制剂,但不同的是它的抑制作用是可逆的。2013年8月在日本上市,该药品由富士药品株式会社和三和化学研究所株式会社研究,商品名分别是Topiloric和Uriadec。
一项随机双盲、平行、阳性对照的Ⅲ期临床研究,用来评估托匹司他的有效性和安全性[9]。203名日本痛风患者(包括高尿酸血症患者)为受试者,使用别嘌醇进行阳性对照(表1)。受试者药物治疗结束时,血清尿酸水平≤6.0 mg/dl的百分率托匹司他给药组为72.4% (71/98例),别嘌醇给药组为73.3%(77/105例)。
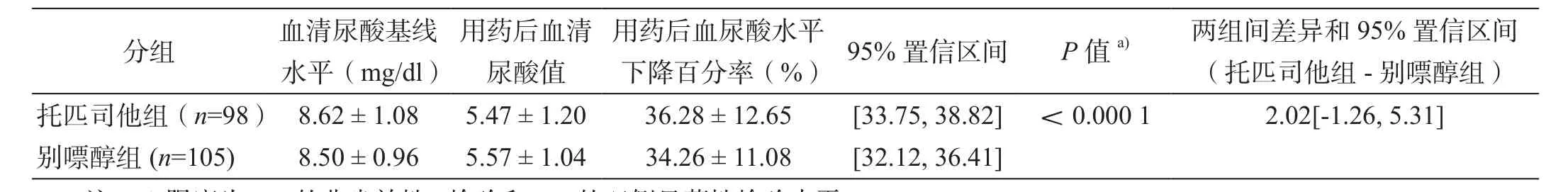
表1 给药结束血清尿酸下降率[9]
受试者是日本痛风患者(含高尿酸血症患者)的一项长期给药研究包括两个时期[9]:适应时期(1~4周)和治疗时期(初始阶段Ⅰ,2周;初始阶段Ⅱ,4周;维持阶段Ⅰ,12周;维持阶段Ⅱ,40周)。受试者每天早晚给药一次。初始阶段Ⅰ20 mg/次(40 mg/d),持续2周;初始阶段Ⅱ40 mg/次(80 mg/d),持续4周。在维持阶段给药60 mg/次(120 mg/d),如果在维持阶段的第14周血清尿酸≤6.0 mg/dl,则将此给药方案延长至40周。如果在维持阶段的第14周,血尿酸值>6.0 mg/dl,则在维持阶段的第18周开始调整剂量为80 mg/次(160 mg/d)持续到第40周。给药26周后(剂量增加到160 mg/d的8周后),若血尿酸值>6.0 mg/dl,在维持阶段的第30周开始给药100 mg/次(200 mg/d)。给药38周后(剂量增加到200 mg/d的8周后),若血尿酸值>6.0 mg/dl,维持阶段的第42周给药120 mg/次(240 mg/d),该剂量至多维持至第58周。如果在维持阶段的第14周和第26周,血清尿酸≤6.0 mg/dl,即使随后血清尿酸增多,也不再增加剂量。若受试者在第2,6,18,30,40周发生痛风性关节炎,可将给药剂量时间段延长1周。给药结束时,患者血清尿酸水平下降率见表2。受试者中,血清尿酸达到≤6.0 mg/dl的比率,在维持阶段的第18周(给药剂量增加前)是70%(110人中有77个),整个治疗过程中是71.9%(121人中有87个)。
托匹司他主要的不良反应是关节痛、头痛、肾损伤、谷草转氨酶/谷丙转氨酶(ALT/AST)增加和a1微球蛋白尿增加[8]。
抑制尿酸形成的除有XO抑制剂外,还有嘌呤核苷磷酸化酶(PNP)抑制剂。它们对尿酸形成途径的抑制作用部位见图1。

表2 托匹司他长期给药研究[9]

图1 尿酸的形成途径及酶抑制剂的作用部位
BioCryst公司研发的ulodesine(BCX4208)是嘌呤核苷磷酸化酶(PNP)抑制剂。理论上,尿酸合成抑制剂的抑制作用越靠前,抑制尿酸合成的作用越强。别嘌醇和非布司他都是抑制尿酸合成过程中终端的酶,而ulodesine作用于嘌呤代谢途径中黄嘌呤和次黄嘌呤的上游,以减少尿酸的生成[10]。ulodesine并不抑制细胞色素P450,也不是一般的转运蛋白的抑制剂,它经肾脏消除,在肝脏中不代谢。数据显示ulodesine与痛风患者常用药物之间有很低的相互作用风险[11]。ulodesine的平行,双盲,随机试验以安慰剂作为对照组(最初受试者血清尿酸>8.0 mg/dl)来证明其有效性(表3)[12]。没有出现与治疗有关的死亡,严重的不良事件是痔出血(40 mg/d ulodesine),不良反应是腹泻,头痛,淋巴细胞数下降,腹部疼痛。ulodesine与别嘌醇联合用药有更好的效果,且显示痛风患者血浆黄嘌呤和次黄嘌呤浓度水平剂量依赖性降低[12]。

表3 ulodesine单独用药有效性及用药22 d后血清尿酸水平改变绝对值[12]
2.3 促进尿酸转换成尿囊素的药物
在动物体内,尿酸可以通过尿酸氧化酶氧化成易溶于水的尿囊素。后者是一种容易排泄的代谢物。但是人体内缺少这种尿酸氧化酶,所以人为地补充这种酶,成为了促尿酸排泄、降血尿酸水平的新策略。拉布立酶(rasburicase)是由Sanofi Synthelabo公司研发的重组尿酸氧化酶,于2001 年6月在英国和德国首次上市,并于2002 年7 月经美国FDA 批准上市,用于治疗和预防具有高危肿瘤溶解综合征的血液恶性肿瘤病人的急性高尿酸血症,尤其适用于化疗引起的高尿酸血症病人[12]。临床研究表明,拉布立酶可快速降低血清中尿酸水平,而且能溶解痛风石,效果优于别嘌醇,更适用于不耐受常规疗法的患者,但该药价格昂贵,半衰期短(仅18 h),需要频繁注射,并且容易引起超敏反应和诱发高铁血红蛋白血症,因此其在临床的使用受到了一定的限制。现在研究中催化尿酸氧化为尿囊素的pegloticase,尽管在Ⅰ期临床研究中表现出潜在降尿酸作用,但是Ⅲ期研究中每两周给药一次组接受18个月治疗后仅有47%的患者达到主要终点(sUA <6.0 mg/dl);另外,使用拉布立酶出现频繁的输液反应(两周用药一次组26%,对照组5%)。pegloticase不良反应发生率高,包括严重的心血管事件,输液反应,免疫原反应[13]。
3 展望
痛风发病率逐年升高,而且发病年龄出现了低龄化,这可能与人们的饮食结构变化密切相关。目前,市场上抗痛风药物虽然耐受性良好,但是品种少、不良反应多,选择受限。改变生活方式,少食用鱼、虾等高嘌呤食物,少饮啤酒可能将成为治疗痛风的新方法。流行病学研究显示乳制品能降低尿酸水平和痛风发作率,而且临床试验也支持低脂乳制品促进尿酸排泄。也许不久将来乳制品的提取物将成为治疗痛风和高尿酸血症毒副作用最少、最有效的药物[14]。
[1] 张源潮, 户中丹, 杨兆文, 等. 治疗痛风新药非布司他的临床研究进展[J]. 世界临床药物, 2014, 35(9): 519-521.
[2] 徐娜, 陈海生. 治疗痛风药物研究进展[J]. 药学实践杂志, 2013, 31(1): 14-18.
[3] Schlesinger N, Alten RE, Bardin T, et al. Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised multicentre, active-controlled, double-blind trials and their initial extensions[J]. Ann Rheum Dis., 2012, 71(11): 1839-1848.
[4] Schumacher HR Jr, Evans RR, Saag KG, et al. Rilonacept (interleukin-1Trap) for prevention of gout flares during initiation of uric acid-lowering therapy: results from a phase III randomized, double-blind, placebo-controlled, confirmatory efficacy study[J]. Arthritis Care Res (Hoboken), 2012, 64(10): 1462-1470.
[5] So A, De Smedt T, Revaz S, et al. A pilot study of IL-1 inhibition by anakinra in acute gout[J]. Arthritis Res Ther, 2007, 9(2):R28.
[6] Sundy J, Perez-Ruiz F, Krishnan E, et al. Efficacy and safety of lesinurad (rdea594), a novel uricosuric agent, given in combination with allopurinol in allopurinol-refractory gout patients: preliminary results from the randomized, double-blind, placebo-controlled, phase 2b extension study [EB/OL]. [2015-05-20]. https://acr.confex.com/acr/2011/webprogram/ Paper23977.html.
[7] Truglio JJ, Theis K, Leimkühler S, et al. Crystal structures of the active and alloxanthine-inhibited forms of xanthine dehydrogenase from Rhodobacter capsulatus[J]. Structure, 2002, 10(1): 115-125.
[8] Okamoto K, Eger BT, Nishino T, et al. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition[J]. J Biol Chem, 2003, 278(3): 1848-1855.
[9] Report on the deliberation results[EB/OL]. (2013-05-08) [2015-04-21]. http://www.pmda.go.jp/files/000153624.pdf.
[10] Hollister AS, Becker M, TerkeltaubR, et al. BCX4208 shows synergistic reductions in serum uric acid in gout patients when combined with allopurinol[EB/OL]. [2015-04-21]. http://www.biocryst.com/PDFs/BCRX_BCX4208_Hollister_ EULAR_2011_Poster.pdf.
[11] BioCryst presents results from its bcx4208 gout program at the annual European congress of rheumatology[EB/OL]. [2015-04-21]. http://www.drugs.com/clinical_trials/biocrystpresents-results-bcx4208-gout-program-annual-europeancongress-rheumatology-13795.html.
[12] Fitz-Patrick D, Drummond W, Pappas J, et al. Effects of a purine nucleoside phosphorylase inhibitor, BCX4208, on the serum uric acid concentrations in patients with gout[EB/ OL]. [2015-04-21]. http://www.biocryst.com/PDFs/BCRX_ BCX4208_Fitz-Patrick_ACR_2010_Poster.pdf.
[13] Liu CY, Sims-McCallum RP, Schiffer CA. A single dose of rasburicase is sufficient for the treatment of hyperuricemia in patients receiving chemotherapy[J]. Leuk Res, 2005, 29(4): 463-465.
[14] Burns CM, Wortmann RL. Gout therapeutics: new drugs for an old disease[J]. Lancet, 2011, 377(9760): 165-177.
[15] Crittenden DB, Pillinger MH. New therapies for gout[J]. Annu Rev Med, 2013. 64: 325–337.
Advances in clinical research of anti-gout drug
ZHANG Li, GUO Yekun, ZHONG Jingfen*
(Shanghai Institute of Pharmaceutical Industry, Shanghai 200437, China)
Gout is a common rheumatologic disorder and non-steroidal anti-inflammatory drugs and colchicine have been currently used for its treatment during its acute attack period. Although these drugs are generally effective, it may be contraindicated especially in patients with concomitant comorbidities while urate-lowering drugs can be used during other periods. In recent years, both xanthine oxidase (XOD) and purine nucleoside phosphorylase (PNP) inhibitors have become a hot topic of research in this field. This paper describes the mechanism of action of the interleukin-1 inhibitor (IL-1β inhibitors), lesinurad, XOD and PNP inhibitors, their clinical effects and adverse reactions.
XOD inhibitors; IL-1β inhibitors; lesinurad; PNP inhibitors
R971.1
A
1006-1533(2015)17-0019-04
钟静芬(1964-),女,研究员,从事心血管和神经系统药物研究。E-mail: zhongjingfen@aliyun. com
2015-04-21)
