无机固体电解质材料的基础与应用研究
2015-09-21陈晓添陶益成许晓雄
黄 祯,杨 菁,陈晓添,陶益成,刘 登,高 超,龙 鹏,许晓雄
(中国科学院宁波材料技术与工程研究所,浙江 宁波 315201)
锂离子二次电池已成功应用于我们生活的各个方面,随着时代的进步和科技的发展,对锂离子电池的要求越来越高[1]。锂离子电池不仅需要具有高的能量密度和功率密度,还需具有使用寿命长、安全性能高等特点,尤其在电动汽车和规模储能领域,对锂离子电池的安全性要求越来越迫切。锂离子电池因过充、内部短路等原因会导致电解液过热,发生起火甚至爆炸事故。此外,电解液与电极材料在充放电过程中会发生副反应,导致电池容量出现不可逆衰减,同时也会带来胀气、漏液等问题。目 前,诸多研究者主要采用在电解液中加入添加剂等方式对有机电解液进行改进,以期解决传统锂离子电池的安全性问题,取得了一定成效,但并不能从根本上消除其安全性问题,因而成为了锂离子电池在动力电池和大容量储能应用方面的障碍。为了彻底解决锂离子电池的安全性问题,一种全新的采用固体电解质的全固态锂电池进入了人们的视线。
采用固体电解质的全固态锂电池工作原理与传统锂离子电池相同[2]。现阶段,锂离子固体电解质材料是全固态锂电池的核心,主要包括聚合物固体电解质和无机固体电解质两类。它们属于在室温或不太高的温度下具有非常高的锂离子电导率、低的电导活化能(<0.5 eV)和非常低的电子电导率的材料,又称为快锂离子导体,其晶格结构具有适宜于离子快速传输的通道或链段,且材料内部存在大量诸如离子空位等缺陷,晶格阳离子及离子缺陷都可以参与离子导电过程[3]。
相比于有机电解液,聚合物电解质具有可塑性强、形状多样化等特点,所以,基于聚合物电解质的锂电池具有可弯曲、易加工等优点,在电子产品市场具有较好的应用前景。但受限于聚合物电解质锂离子电导率低、易析晶等缺点,在动力和储能领域,聚合物电池的综合性能还有很大提升空间[4]。然而,无机固体电解质具有明显的特点和优势,主要包括以下几个方面:① 无任何液体成分,不可燃,可有效避免燃烧和泄漏等安全问题;② 机械加工性能好,可以根据要求制作成所需形状;③ 组装电池时,固体电解质兼具传导锂离子与正负极隔膜的双重作用,可简化电池结构;④ 采用无机固体电解质的固态锂电池工作温度范围宽,适用温度范围在–70~500℃;电池工作电压高,理论上比传统锂离子电池可以具有更高的能量密度;⑤ 固体电解质化学稳定性和电化学稳定性好,电解质与电极间的固固副反应慢,能很好地减缓循环过程中的容量衰减,提升电池循环寿命。此外,固体电解质在锂硫电池和锂空电池中作为隔膜材料都具有很好的应用前 景[2, 5-7]。
无机固体电解质材料主要可分为氧化物体系和硫化物体系两类。室温锂离子电导率高的氧化物体系电解质包括(反)钙钛矿型结构、NASICON型结构、LISICON型结构和石榴石型结构。与O2-相比较,硫的电负性更小,因此对锂离子的束缚力就小,且S2-离子半径较O2-大,导致晶格结构中的离子迁移通道会大,更有利于锂离子的快速迁移。硫化物体系电解质主要包括 Li2S-P2S5基二元硫化物和Li2S-P2S5-MeS2(Me=Si、Ge、Sn等)基三元硫化物固体电解质材料。针对上述锂快离子导电材料的基础及应用研究,本文对当前最具备应用前景的无机固体电解质材料——氧化物体系中的NASICON型结构和石榴石型结构固体电解质、硫化物体系固体电解质进行综合论述,对其发展历程、材料特点、制备改性和应用前景进行了详细介绍,并指出其未来应用过程中的挑战和趋势。
1 NASICON结构型固体电解质
1.1 NASICON结构型固体电解质概述
NASICON(sodium super ion conductors)结构类型快离子导体是一类被广泛研究的固体电解质材料,该类型材料的晶体结构于 1968年被第一次研 究[8]。1976年,Goodenough和 Hong[9]报道了Na3Zr2Si2PO12的合成,即目前通常被称为NASICON的Na+离子导电材料。这类化合物的分子式一般为M[A2B3O12],其中M、A、B分别代表一价、四价和五价的阳离子,其骨架结构是由AO6八面体与BO4四面体共同形成,属于R3c空间点群[10]。每个 AO6八面体与 6个 BO4四面体相连接,每个BO4四面体与4个AO6八面体相连接,这些多面体通过相互接触的顶角氧原子相连,组成三维互连的骨架结构,形成平行于c轴的离子传输通道,其结构及离子传输示意图如图1所示。在这种结构中,M+导电离子可以占据两种填隙位置,这些位置被称之为MI(八面体空隙,蓝球所占位置)和MII(四面体空隙,黑色中空方形位置)。由于MI位的势能比 MII位低,故 MI位被 M+全部占满,通常 MII位则未被占据,所以,在 NASICON结构中,M+的迁移路径有两种:一种是通过 MIMII瓶颈的MI→MII跃迁,另一种是通过 MIIMII瓶颈的MII→MII跃迁。M+导电离子通过瓶颈从一个位置迁移到另一个位置时,存在[A2B3O12]-骨架收缩和M+迁移的协同运动,传输通道与迁移离子的半径达到一定的匹配程度才能有利于 M+迁移,而瓶颈的大小取决于骨架离子[A2B3O12]-的大小,因此,NASICON结构化合物的离子导电性能会随着骨架离子的组成而改变[11-12]。提高NASICON结构固体电解质离子电导率的前提条件是离子通道与传输离子半径大小必须匹配,骨架结构对迁移离子的束缚 作用弱,另外,材料的结构稳定且致密度高[13-14]。
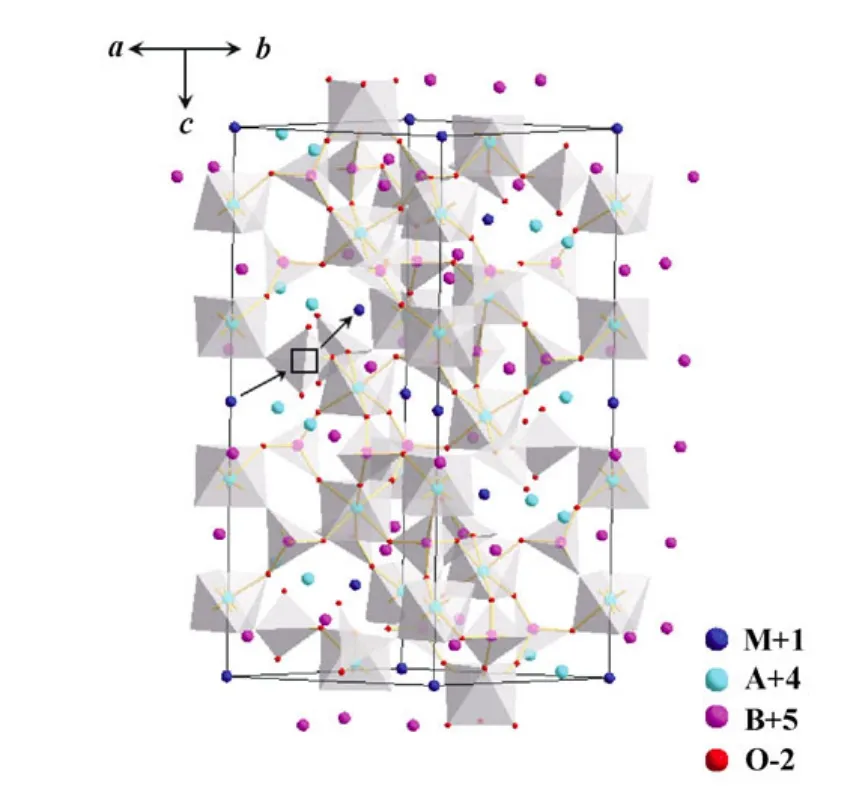
图1 NASICON结构示意图 Fig.1 Structure schematic of NASICON type electrolyte notes
将 Na3Zr2Si2PO12中的 Na+换成 Li+,就成为锂离子电池固体电解质,但是直接取代得到的Li3Zr2Si2PO12锂离子电导率很低,较Na3Zr2Si2PO12的钠离子电导率低约 3个数量级,原因即为Na3Zr2Si2PO12结构中原本适合 Na+迁移的传输通道尺寸相对Li+太大,不适合Li+的迁移,解决方案是用不同大小的离子对结构骨架离子进行取代,进而改变传输通道尺寸[15]。其中,Zr4+可以被Ti4+、Ge4+、Hf4+、V5+或 Sc3+取代,取代后的化合物仍具有NASICON结构,而材料的锂离子电导率得到大幅度提高[16-17]。由于该类型结构锂快离子导体具有结构性质稳定、合成方便等特点,其作为高离子电导固体电解质将具有很好的应用潜力,然而其室温电导率还有待进一步提升,因此提高 NASICON结构类型材料室温锂离子电导率成为了研究开发的重点。
众所周知,大部分无机固体电解质为多晶材料,晶粒和晶界电导对无机固体电解质总电导的贡献构成了其宏观导电特性,而且晶界的贡献在一定条件下还会占主导地位,显著地影响材料整体的离子导电性能。NASICON结构类型的陶瓷锂离子电解质的室温晶粒离子电导率可以超过10-3S/cm,但晶界电导率要低1~2个数量级,使得NASICON结构类型陶瓷电解质材料的总离子电导率不高。经过多年 的发展,NASICON结构类型固体电解质的室温总离子电导率已经有了明显提高,其改性思路也主要是通过晶粒和晶界的共同作用来实现的。采用尺寸合适的低价离子取代骨架结构中的高价离子,如Al3+、Ga3+对 A位取代,Si4+对 B位取代。在没有改变晶相结构的前提下,低价离子的引入一方面可以改变离子传输通道的大小,另一方面由于电荷平衡可以引入更多可迁移的 Li+,进而从两方面影响材料的晶粒离子电导率[18-20],这种方法已被广泛采用。在晶界调控方面的研究,首先是优化制备方法,普通固相反应法难以制得颗粒尺寸均匀、物相单一的产物,共沉淀法及溶胶-凝胶法被用来合成产物,产物均匀性和物相纯度得到明显改善[21-23];其次,使用传统固相反应法,即使在很高的温度下也难以形成结构致密的产物,而放电等离子烧结 SPS (spark plasma sintering)和机械化学合成法等新型制备工艺可以提高产物致密度,甚至达到大于99% 的相对密度,室温离子电导率也因此得到提高[24-25]。为了通过提高材料致密度而达到改善晶界效应的目的,在样品成型前加入不发生化学反应的低熔点含锂化合物,如Li3PO4、Li3BO3等,在烧结过程中填充在晶粒间空隙中,改善界面接触,进而提高致密度,同时还利于提高晶界处的导电 Li+浓度,最终达到提高材料总离子电导率的目的[26]。采用退火处理等热处理方法也可以改善陶瓷样品的颗粒形貌,消除残余应力,提高材料的离子电导率。另外,由于微晶材料是由玻璃母体析晶形成玻璃与晶相的复合物而得到,会有玻璃相存在于晶界处起到协同、调控作用,能有效地降低晶界电阻,提高材料的致密度和总离子电导率。
在NASICON结构类型锂离子导电材料中,研究最多的是 LiM2(PO4)3(M=Zr,Ti,Ge,Hf)材料,常见的有主要是 Li2O-Al2O3-TiO2-P2O5(简称为LATP)和Li2O-Al2O3-GeO2-P2O5(简称为LAGP)两个体系,相对而言,M为Ti的体系在NASICON结构类型锂离子导体中具有最佳的离子导电通道尺寸,故离子电导率相对更高[27]。
1.2 LATP基锂离子固体电解质
从制备方法上来看,对LATP体系的研究不再局限于传统的固相反应法,并取得了很好的进展。Kotobuki等[21, 28]分别采用 Al(C3H7O)3和 Al(NO3)3作为Al源,通过溶胶-凝胶法制备了Li1.5Al0.5Ti1.5(PO4)3陶瓷。采用两种不同磷源虽然均能得到LATP体系的产物,但Al(NO3)3作为Al源时产物中含有AlPO4杂相,并成为阻塞层阻碍锂离子传输。结果显示,不同Al源造成了晶界离子电导率的显著不同,同时晶粒离子电导率也有变化,引起总离子电导率的不同。Yamamoto等[29]采用溶胶-凝胶法制备了致密的Li1.4Al0.4Ti1.4Ge0.2(PO4)3材料,离子电导率可达1.29×10-3S/cm,其致密度达到 94.4%。Xu等[24]通过SPS方法合成了Li1.4Al0.4Ti1.6(PO4)3陶瓷,不仅大幅降低了烧结温度,而且制得样品的晶粒尺寸达到纳米量级,且相对密度极高,室温总离子电导率达到1.12×10-3S/cm。同传统的固相法制备的同组分样品相比,其晶界离子电导率提高了约2倍,这说明当颗粒降低至纳米尺寸,致密度得到极大改善,提高了锂离子在晶界处的传导。Morimoto等[25]采用机械研磨法制备了Li1.3Al0.3Ti1.7(PO4)3材料,样品室温离子电导率达到10-4S/cm数量级。Wen等[6]也用相同的方法制备了Li1.4Al0.4Ti1.6(PO4)3样品,室温离子电导率达到5.16×10-4S/cm,并指出电学性能的提高可以通过降低晶粒尺寸来实现。这些方法虽然在较低烧结温度下实现了样品的微晶化,但是要实现电解质材料的规模化制备仍具有很大困难。
从掺杂改性晶粒电导来看,主要是针对Ti位和P位的掺杂取代工作。Aono等[15]早在1990年前后就对LiTi2(PO4)3材料进行了有启发性的研究工作。分别对其 Ti位和 P位进行不同离子种类的取代研究,发现采用 Al3+和 Sc3+掺杂后的样品Li1.3Al0.3Ti1.7(PO4)3和 Li1.3Sc0.3Ti1.7(PO4)3室温总离子电导率最高可达7×10-4S/cm;而采用Si4+掺杂取代P5+后样品Li1.3Ti2(P0.9Si0.1O4)3的室温离子电导率达到3.2×10-4S/cm,但XRD图中发现存在杂相,说明引入的 Si4+并不能完全进入 P5+位进行取代。Johnson等[30]对LiTi2(PO4)3材料进行了共取代研究,制备了Li1.5Al0.3Ti1.7Si0.2P2.8O12材料,分别采用Al3+和Si4+对部分Ti4+和P5+进行取代。制备过程中发现取代后可以降低孔隙率,并增大玻璃液的流动性。然而Li1.5Al0.3Ti1.7Si0.2P2.8O12体系玻璃在后续热处理过程中容易出现开裂现象,于1000℃下热处理样品室温电导率可达2.77×10-4S/cm。
此外,利用空间电荷效应,通过复合其它相的方法来改善材料性能也有报道。Kumar等[31]加入少量高介电相BST和低介电相Al2O3与LATP玻璃陶瓷进行复合,尝试利用空间电荷效应来提高离子电导率。结果显示,在晶界处的介电相存在吸附 Li+的作用,其中高介电相 BST的效果比低介电相Al2O3明显。然而进行介电相复合的样品电导率低于没有进行介电相复合的样品,原因是介电相在烧结时过度生长,出现阻塞效应,导致电导率降低。若能有效降低介电相颗粒尺寸,便可减小阻塞效应影响。
1.3 LAGP基锂离子固体电解质
虽然LATP体系固体电解质室温电导率要高于LAGP体系固体电解质,但 Ti4+的稳定性要弱于Ge4+,在较低电势条件下易发生还原反应生成Ti3+,离子电导率会限制降低,阻碍了其在锂二次电池中的应用。更为重要的是,LAGP电化学窗口在1.0~6.0 V间具有稳定的电化学性能[32],因此要提高电化学窗口范围,NASICON型结构锂离子固体电解质材料中应当尽量减少Ti的含量。对LAGP体系性能的研究同样主要是通过工艺改进和组分优化两个方面,从而达到提高晶粒离子电导率或者晶界离子电导率,进而提高总离子电导率。
Fu等[33]详细研究了 Li1+xAlxGe2-x(PO4)3系列玻璃陶瓷材料,制备了 x=0、0.2、0.4、0.6和 0.8组分的样品,并对性能进行表征,结果表明x=0.6体系的性能最佳。进一步由晶化温度和玻璃化温度之差比较发现x=0.5体系稳定性更好。当x=0.5~0.6时,材料具有高的室温电导率和低的激活能 Ea。何坤 等[34]制备了 Li1+xAlxGe2-x(PO4)3(x=0~0.7)体系玻璃陶瓷,探究了 Al2O3对样品性能的影响。结果表明,x=0.5时,体系离子电导率最高,可达5.8×10-4S/cm。当x>0.5时,样品中出现AlPO4杂相,会聚集在晶界处堵塞离子传输通道,导致离子电导率下降。Fu等[35]随后研究了不同三价离子取代即Li1.2M0.2Ge1.8(PO4)3(M= Al,Ga,Y,Dy,Gd 和La)体系玻璃陶瓷的导电性。结果表明,通过改变取代离子种类,可调整离子传输通道尺寸,从而影响离子电导率。Al3+和 Ga3+取代样品有较高的离子电导率,其它离子由于离子尺寸匹配性不佳,造成了晶格结构的畸变,使离子电导率下降,性能劣化。Takashi等[36]鉴于LAGP的稳定性和LATP的良好室温离子导电性,提出将两种体系结合起来,同时加入Si4+对P5+的部分取代,制备了Li1+x+yAlx(Ti2-yGey) P3-zSizO12玻璃陶瓷,并将其应用于电池中。结果表明,样品相比LATP样品电化学稳定性得到了改善,但组装电池后对Li金属的稳定性没有明显改善。这些研究表明,Al和Ga的Ge位掺杂是一种可以提高离子电导率的有效方法,其原因就在于Al和Ga掺杂改变了晶格中锂离子迁移通道的大小,对晶粒离子电导率的提高有利。
另一方面,针对LAGP体系晶界效应的调控,研究者们提出了诸多思路。Xu等[32]研究了锂源过量对LAGP玻璃陶瓷样品结构与性能的影响,制备了 Li1.5Al0.5Ge1.5(PO4)3-xLi2O(x=0,0.05,0.075,0.1和0.2)体系玻璃陶瓷。研究表明,锂过量有助于提高样品的致密度,但锂过量程度过大则会使晶粒异常长大,晶粒与晶粒之间出现间隙,导致致密度下降。x=0.05时,样品离子电导率达到7.25×10-4S/cm,此时具有最高的晶粒、晶界离子电导率和总离子电导率。Jadhav等[37]探究了 B2O3添加剂对样品性能的影响,研究表明B2O3会降低样品的晶化温度,并增进晶界锂离子扩散,进而增大总离子电导率。当B2O3添加量为 0.05 %(质量分数)时,样品离子电导率达到6.9×10-4S/cm。
此外,针对LAGP体系制备工艺的改进工作也有很多报道。Cruz等[38]制备了 Li1.5Al0.5Ge1.5(PO4)3体系玻璃陶瓷。他们确定了 Li1.5Al0.5Ge1.5(PO4)3玻璃陶瓷的最快形核温度及最佳晶化温度,通过改进热处理工艺来提升样品致密度,解决了样品中常见的孔洞缺陷。但是这种方法并未从根本上提高离子电导率。这是因为样品晶界含量过高,对离子传输起到阻碍作用,故降低了材料的总离子电导率。Thokchom 等[39]分别采用熔融-淬冷法和烧结法两种方法制备 Li1.5Al0.5Ge1.5(PO4)3玻璃陶瓷并进行了系统研究,结果显示烧结法样品相对密度高于熔融-淬冷法样品 4.3%,然而,熔融-淬冷法制得的样品室温晶粒电导率要高于烧结法样品。
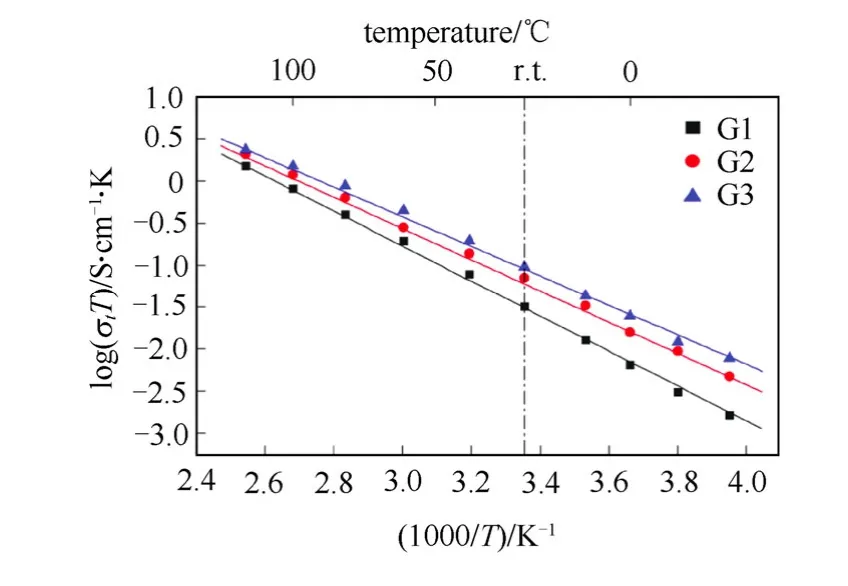
图2 三代Li1.5Al0.5Ge1.5(PO4)3基微晶材料离子电导率随温度的变化 Fig.2 The temperature dependence of ionic conductivities of three generations in Li1.5Al0.5Ge1.5(PO4)3-based microcrystallines
近年来,中国科学院宁波材料技术与工程研究所在NASICON结构固体电解质材料的制备和研究方面也取得了不错的进展。目前已经实现了第三代(G3)的Li1.5Al0.5Ge1.5(PO4)3基微晶材料的百公斤级生产,其量化制备的材料室温总离子电导率可以达到 6.21×10-4S/cm,相对密度超过 97%,一致性良好。图 2为我们制备的三代(第一代 G1、第二代G2和第三代G3)Li1.5Al0.5Ge1.5(PO4)3基微晶材料离子电导率随温度的变化关系的对比图。通过Arrhenius公式拟合,可得对应的激活能 Ea。表 1给出了G1、G2、G3的室温离子电导率和对应的激活能以及相对密度。图3为三代Li1.5Al0.5Ge1.5(PO4)3基微晶材料的SEM图,可以看出,G3材料晶粒形貌更加均一,孔洞结构逐渐减少,表现出更加优异的形貌特征。

表1 三代Li1.5Al0.5Ge1.5(PO4)3基微晶材料室温离子电导率、激活能和相对密度 Table 1 Room-temperature ionic conductivity, activation energy and relative density of three generations in Li1.5Al0.5Ge1.5(PO4)3-based microcrystallines

图3 三代Li1.5Al0.5Ge1.5(PO4)3基微晶材料SEM图 Fig.3 SEM images of three generations in Li1.5Al0.5Ge1.5(PO4)3-based microcrystallines
综上所述,NASICON结构体系快离子导体材料具有高的室温离子电导率、化学稳定性良好、电化学窗口宽,适合作为固体电解质应用于高电压全固态锂电池,同时是一种理想的固体隔膜材料,具有良好的应用前景。未来发展方向将会主要集中在优化制备工艺,提高晶界和晶粒离子电导率,提升致密度来进一步改善室温离子电导率,并不断拓展其应用领域。
2 石榴石结构型固体电解质
在理想石榴石 X3Y2(SiO4)3(X=Ca,Mg,Fe等;B=Al,Cr,Fe等)中,X和Y分别是氧离子的八配位和六配位离子。可以看出,石榴石结构中含有八配位、六配位和四配位的阳离子,且各种阳离子处于不同的价态,因此,用碱金属、碱土金属、稀土金属和过渡金属离子取代X、Y或者Si离子成为可能。
2.1 石榴石结构型固体电解质结构特征
含Li石榴石材料早在1969年就被Kasper发现[40],当时研究的材料为 Li3Ln3M2O12(M=W,Te)。随后 Mazza[41]和 Hyooma等[42]研究了La2O3-M2O5-Li2O(M=Nb,Ta)的三元相图,并分别利用粉末 XRD和单晶 XRD研究了石榴石Li5La3M2O12(M=Nb,Ta)的结构。Mazza认为,Li5La3M2O12(M=Nb,Ta)的结构和石榴石结构紧密相关,空间群为 Ia-3d,晶胞参数 a=12.8 Å(1Å=0.1 nm)。图 4给出了 Li5La3M2O12(M=Nb,Ta)1/8单胞结构图,其中,La占据X位置(1/8,0,1/4),Nb或 Ta占据Y位置(0,0,0),3个Li占据 Si的四配位位置(3/8,0,1/4),剩下2个Li占据八配位位置(1/8,1/8,1/8),而这个八配位位置在理想石榴石结构中是不被占据的。Hyooma等则认为Li5La3M2O12(M=Nb,Ta)的结构为更低对称性的非中心对称I213空间群结构,其中金属阳离子有两个独立的晶体学位置,如La(x,0,1/4),Nb或Ta(0,0,0)和(1/4,1/4,1/4),而Li则占据两个不同的八面体位置。Cussen[43]利用中子粉末衍射对Li5La3Ta2O12进行了研究,进一步确定了其空间群为Ia-3d,并且发现Li分别占据八面体和四面体位置,这些位置的连接构成了Li离子迁移的三维通道。虽然被Li离子占据的八面体位置并不多,但却是石榴石结构中Li离子迁移的主要原因。Cussen还发现,高温下Li5La3Ta2O12和室温下Li5La3Nb2O12的Li占位都具有类似的特征。
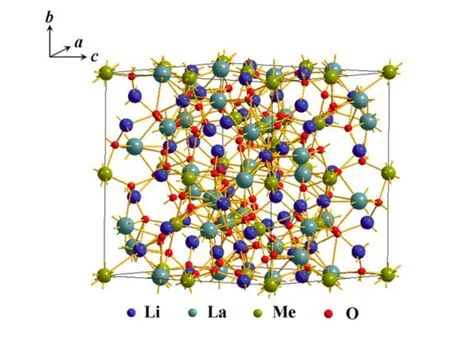
图4 Li5La3Me2O12(Me=Nb,Ta)的结构示意图 Fig.4 Structure schematic diagram of Li5La3Me2O12(Me=Nb, Ta)
2.2 石榴石结构型固体电解质研究现状
德国基尔大学的Thangadurai和Weppner等[44]在2003年前后首先发现了Li5La3M2O12(M=Nb, Ta)可以作为锂离子无机固态电解质材料。Li5La3M2O12(M=Nb,Ta)表现出典型的纯锂离子导电的特征,且具有很好的电化学稳定性和化学稳定性,且纯Li5La3M2O12(M=Nb,Ta)在室温下的电导率约为10-6S/cm,激活能分别为 0.43 eV(Li5La3Nb2O12)和0.56 eV(Li5La3Ta2O12)。研究表明,Nb系总电导率要略高于Ta系,但是Ta系与熔融Li反应的稳定性要高于Nb系。
2.2.1 La位掺杂
碱土金属掺杂取代La的Li5La3M2O12(M=Nb,Ta)研究表明,这种掺杂方法可以很好地降低晶界电阻,提高总锂离子电导率[45-46]。在碱土金属掺杂的Li5La3Nb2O12中,合成样品中会出现微弱的钙钛矿杂相结构 La2LiNbO6[46]。Li6ALa2Nb2O12(A=Ca,Sr,Ba)的高温晶粒电导率相比,Ba掺杂后的晶粒电导率最大,且拥有最低的激活能0.44 eV。利用等效电路拟合,表 2给出了 22℃空气中测量的Li6ALa2Ta2O12(A= Sr,Ba)阻抗拟合数值,其中Rbulk和Cbulk分别是等效电路中晶粒电阻和晶粒电容,而Rgb和Cgb分别是等效电路中晶界电阻和晶界电容,Cel为接触电容。可以看出,掺杂Sr和Ba后,晶界电阻Rgb大幅降低,分别约为晶粒电阻的1/5和2/5。

表2 22℃空气中测量的Li6ALa2Ta2O12(A= Sr,Ba)阻抗拟合数值[45] Table 2 The impedance fitting parameters of Li6ALa2Ta2O12(A= Sr, Ba) measured at 22℃[45]
2.2.2 M 位掺杂
除了La位的碱土金属掺杂外,M(M=Nb, Ta)位的掺杂也开始被陆续报到。目前研究最多的是Zr取代M,它具有迄今为止含锂石榴石结构材料最佳的电导率,纯 Li7La3Zr2O12材料的室温总电导率为3.0×10-4S/cm,激活能 0.30 eV 左右[47]。和其它锂离子固体电解质材料相比,Li7La3Zr2O12在100 ℃以下具有更高的总离子电导率,仅次于Li1.3Al0.3Ti1.7(PO4)3,如图5所示。其高锂离子电导率要归因于以下几点:①Zr离子半径要大于M 离子,使得传导锂离子的三维骨架增大;②锂离子迁移浓度的提高;③锂离子在晶格中同其它离子的化学作用减弱;④陶瓷样品的致密度较高(相对密度达到92%)。

图5 氧化物锂离子固体电解质和Li3N锂离子固体电解质总电导率的对比[47]Fig.5 The temperature dependence of ionic conductivity of oxide lithium electrolytes and Li3N electrolyte[47]
除了碱土金属在La位掺杂和Zr在M位掺杂外,碱金属的La位掺杂和更低价的金属在M位的掺杂也有报道,其效果是间接地提高了迁移 Li+的含量,为电导率的提高奠定了基础。如K掺杂后的Li5.5La2.75K0.25Nb2O12和 In掺杂后的 Li5.5La3Nb1.75In0.25O12[48],均表现出比 Li5La3Nb2O12要高的电 导率。
2.3 Li7La3M2O12(M=Zr, Sn)系固体电解质
Weppner等[47]于2007年率先报道了Zr系石榴石Li7La3Zr2O12(LLZ)结构的固体电解质陶瓷,报道的电解质材料的室温总电导率为3×10-4S/cm,这也是目前报道的电导率最高的一类石榴石结构材料。Weppner等给出LLZ的XRD图谱与先前报道[43]的Li5La3M2O12(M=Nb,Ta)立方石榴石相(空间群 Ia-3d)结构吻合。Awaka等[49]则使用了 Li2CO3作为Li源,利用金坩埚、采用熔盐法制备LLZ单晶,利用单晶 X射线衍射方法制备了空间群为I41/acd的LLZ晶体,这也是首次报道的四方结构的LLZ晶体。然而,与立方结构的LLZ晶体相比,四方结构LLZ晶体表现出较差的电导率性能。四方结构LLZ晶体的室温体电阻σb=1.63×10-6S/cm,室温晶界电阻σgb=5.59×10-7S/cm。文中对电导率性能的急剧降低提出了可能的原因:Li原子和 Li空位完全有序的排列在四方 LLZ晶体中四面体位置和八面体位置。然而,对于立方LLZ晶体来说,则表现出在四面体位置和八面体位置更为复杂无序的 Li原子和 Li空位。由此可以得出结论,完全有序的Li原子-空位对抑制了Li位之间的离子跳跃,同时由于和立方石榴石结构相比,四方LLZ晶体的等价Li位减少,最终导致了低Li离子电导率。
LLZ为什么在室温下出现了两种不同的相结构且对应的电导率差别很大。Wepper等[50]对这个问题进行了比较深入的分析。他们采用两种不同的烧结方法:方法一是合成阶段采用陶瓷坩埚,后期采用密封Pt囊,用以生成单晶材料;方法二是全过程采用Pt坩埚来制备晶体材料。利用方法一制备材料的元素分析显示 Al元素的存在,这是因为使用的Li2CO3和LiOH在制备材料过程中会形成少量的融熔相,从而溶解了刚玉坩埚中的部分 Al2O3。这种材料在测试的变温 XRD温度范围内均表现出立方相结构,空间群Ia-3d。利用方法二制备得到的材料在测试变温 XRD温度范围内表现出从四方结构向立方结构转变的过程,其相变温度在100~150 ℃。相对于导电性能较差的四方结构来说,通过少量Al3+的加入可以将 LLZ高温立方相结构保持到室温。这是因为:①立方结构材料在三维方向上具有各向同性的Li离子扩散路径;②相隔紧密的Li位和离域Li的作用保证了Li离子扩散的容易和速度;③Li空位较多,这很有可能是由于等价取代造成的,如 Al3+3Li+。Adams等[51]利用原位变温XRD测试和相关模拟研究,发现了类似的现象,即四方相结构的材料表现出有序的锂离子重排,大部分的Li完全占据16f和32g的八面体位置,两类四面体位置中8a位置被完全占据,16e八面体位置则表现出略高的能量位置,从而影响了其电导率的提高。立方相结构的研究表明,完全被占据的四面体位置和低占据率的扭曲的八面体位置实现 Li+的重排,表现出无序特征,有利于离子电导率的提高。
关于LLZ掺杂的研究也比较多,逐渐将室温总锂离子电导率从 3.0×10-4S/cm 提高到 1×10-3S/cm左右。首先,人们通过低价离子的掺杂取代,提高Li的化学计量,从而在锂离子电导率的提高方面取得了一定效果。如 Murugan等[52]通过选择 Y稳定ZrO2(3%的YSZ)作为原料,制备了Li7.06La3Y0.06Zr1.94O12,其室温总锂离子电导率达到了8.1×10-4S/cm。随后,科研人员开始采用高价离子掺杂取代,尤为Ta5+和Nb5+掺杂取代Zr4+研究最多。Ohta等[53]利用Nb掺杂 Li7-xLa3(Zr2-x,Nbx)O12(x=0~2),在 x=0.25 时获得总锂离子电导率为8×10-4S/cm,此时的激活能也达到最低,为30 kJ/mol;Allen等[54]则采用共沉淀法制备前驱粉体,并利用热压的烧结方法制备了Ta掺杂后的 Li6.75La3Zr1.75Ta0.25O12,获得总锂离子电导率为 8.7×10-4S/cm,激活能为 0.22 eV;Goodenough 等[55]研 究 了 Li7-xLa3Zr2-xTaxO12(x=0~1),采用氧化铝作为坩埚,发现当0.4≤x≤0.6时,Li7-xLa3Zr2-xTaxO12具有较好的锂离子电导率。尤其在x=0.6时,具有最佳的室温锂离子电导率,为1×10-3S/cm,此时材料中含有少量的Al元素,而正是这些Al元素以无定形相存在于晶界处,起到了烧结助剂和阻止Li元素挥发的作用。Ta5+和Nb5+掺杂取代Zr4+的研究表明,Li的化学计量一定程度地降低可以提高锂离子电导率,因此更高价态的W6+和Te6+掺杂取代Zr4+的工作也开始被研究。Murugan 等[56]制备了 W 掺杂的 Li7−2xLa3Zr2−xWxO12(x= 0.3,0.5),发现在 x=0.3时,可获得室温总锂离子电导率为7.89×10-4S/cm的固体电解质材料;Murugan 等[57]还利用 Te6+掺杂取代 Zr4+,在 1100 ℃下成功烧结制备了Li6.5La3Zr1.75Te0.25O12,其室温总锂离子电导率为 1.03×10-3S/cm,并指出获得LixLa3M2O12固体电解质材料最佳室温锂离子电导率下的锂离子化学计量x=(6.4±0.1)。
3 硫化物锂离子固体电解质
3.1 硫化物锂离子固体电解质概述
3.1.1 硫化物锂离子固体电解质的分类
硫化物固体电解质可分为两类,一类是Li2S-SiS2体系,另一类是Li2S-P2S5体系。过去对硫化物固体电解质的研究主要是对 Li2S-SiS2体系的研究,但研究结果表明,Li2S-SiS2体系固体电解质的离子电导率不高,虽然可通过添加LixMOy(M=Si,Ge,P)等锂盐提高材料的离子电导率[58],但提升空间有限,其化学性能、物理性能与Li2S-P2S5体系相比,存在较大差距。
Li2S-P2S5体系硫化物固体电解质按照组成可分为二元硫化物固体电解质(主要由Li2S和P2S5两种硫化物组成的固体电解质)和三元硫化物固体电解质[主要由 Li2S,P2S5和 MS2(M=Si,Ge、Sn 等)三种硫化物组成的固体电解质];按物相可分为玻璃类硫化物固体电解质、玻璃陶瓷类硫化物固体电解质和陶瓷类硫化物固体电解质。由于Li2S-P2S5体系硫化物固体电解质的离子电导率较高、电化学窗口宽、电子电导率低,是目前研究最多的硫化物固体电解质。
3.1.2 硫化物锂离子固体电解质的结构特征
大多数硫化物电解质的结构类似于 Kanno 和 Murayama[59]于 2001年发现的基于 Li4GeS4-Li3PS4固溶体系的thio-LISICON结构。根据不同超晶格的衍射数据,固溶体 Li4-xGe1-xPxS4(0<x<1.0)的thio-LISICON结构分为三个部分:区域Ⅰ为 0<x≤0.6;区域Ⅱ为0.6<x<0.8;区域Ⅲ为0.8≤x<1.0。其中区域Ⅰ(0<x≤0.6)对应于单斜超晶格 a×3b×2c结构,这与其正交 Li4GeS4母相晶格相关;区域Ⅱ(0.6<x<0.8=对应于单斜超晶格 a×3b×3c结构;区域Ⅲ(0.8≤x<1.0=当 x=0.8时,其结构由单斜超晶格a×3b×2c构成。这三个区域的不同单斜超晶格结构对应于不同的阳离子有序度。研究结果显示,区域Ⅱ离子电导率要优于区域Ⅰ和区域Ⅲ,当x=0.75,所制备的硫化物电解质室温离子电导率达到 2.2×10-3S/cm。
Hayash等[60]通过高能球磨法制备了 80%Li2S- 20%P2S5(摩尔分数),75%Li2S-25%P2S5二元硫化物电解质材料。通过 XRD图谱的比较,80%Li2S-20%P2S5在240 ℃烧结所得产物的结构与Kanno 和 Murayama 提到的 thio-LISICON Ⅱ区结构极为类似,其室温离子电导率达7.2×10-4S/cm;相应地,75%LI2S-25%P2S5在230 ℃烧结所的产物的结构类似于 thio-LISICON Ⅲ区结构,其室温离子电导率为 2.8×10-4S/cm。这也证明了thio-LISICON Ⅱ结构具有比thio-LISICON Ⅲ结构更高的室温离子电导率。Mizuno等[61]于2005年制得 70%Li2S-30%P2S5室温离子电导率达 3.2×10-3S/cm的电解质材料,远高于其它报道的二元硫化物电解质材料的离子电导率。Raman光谱显示,其晶体结构不同于任何 thio-LISICON系列的电解质材料,对于其晶体结构的表征还需进一步研究。
硫化物电解质中最重要的快离子导体是Li7P3S11晶体,其它种类的快离子导体可认为是由该晶体衍生而出。研究结果发现,当温度达到420 ℃时,Li7P3S11可分解为 Li4P2S7和 β-Li3PS4两种快离子导体。Raman光谱显示[61],Li3PS4的衍射峰在419 cm-1处,Li4P2S7和 Li4P2S6的衍射峰分别在 406 cm-1和382 cm-1。当改变组分和热处理温度时,Li4P2S7易发生分解反应:Li4P2S7→Li4P2S6+1/8S8,S8的导电性极差,Li4P2S6属于慢离子导体。因此,制备硫化物固体电解质时须严格控制析晶条件[62]。图6和图7分别给出了β-Li3PS4和Li7P3S11的结构图。其中,β-Li3PS4属于正交晶系,由 PS4四面体和 LiS4四面体构成骨架结构,而 Li7P3S11属于三斜晶系,除了PS4四面体和LiS4四面体,还有S4四面体、LiS3平面和LiS5四面锥结构,从而构成了其骨架结构。
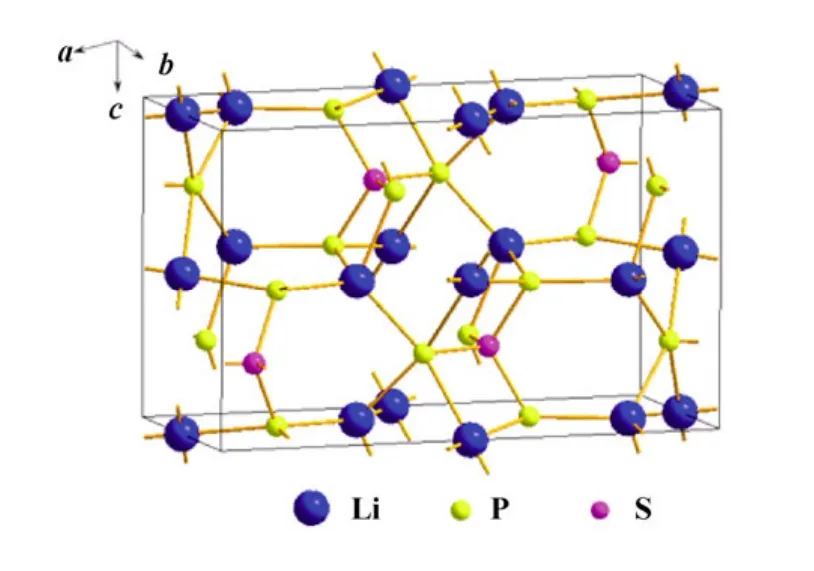
图6 β-Li3PS4的晶格结构示意图 Fig.6 The framework strucure of β-Li3PS4lattcie cell
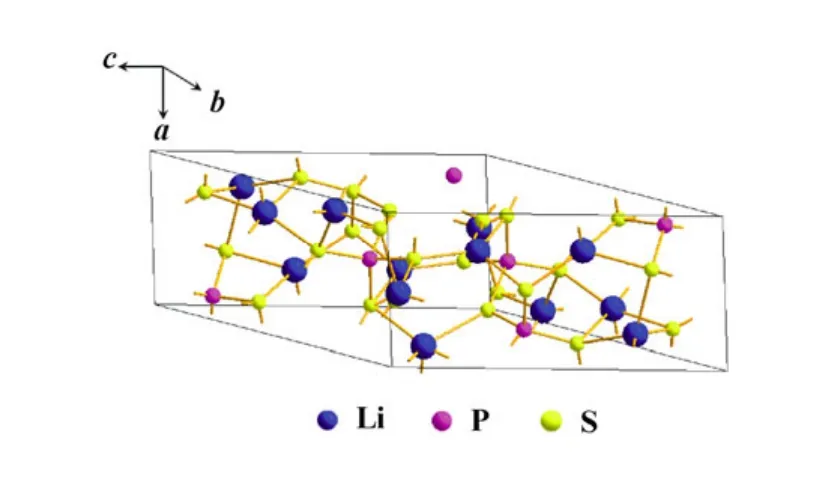
图7 Li7P3S11的晶格结构示意图 Fig.7 The framework strucure of Li7P3S11lattice cell
由Kamaya等[63]发现的一种新的超锂离子导体——Li10GeP2S12,室温离子电导率高达10-2S/cm,事实上,这种固体电解质材料就是 Kanno 和 Murayama等研究的Li4-xGe1-xPxS4体系中x=2/3(区域Ⅱ)并采取不同的制备工艺所得。Li10GeP2S12是由(Ge0.5P0.5)S4四面体、PS4四面体、LiS4四面体以及 LiS6八面体构成的三维网状结构,(Ge0.5P0.5)S4四面体和LiS6八面体二者共棱且形成沿c轴的一维长链,即构成了该框架结构沿 c轴方向的一维 Li离子迁移通道,如图8显示的Li10GeP2S12晶格结构所示,一维 Li离子迁移通道由 LiS4四面体的 16h和8f位置构成,二者共棱且形成一维四面体链,这些链由LiS4四面体共顶点相连。中子衍射分析表明:锂离子在 16h和 8f位置的热振动是高度各向异性的,各向异性热位移表明,锂离子从16h和8f位置移向2个16h位置以及16h和8f位置的间隙位置,这清楚地表明沿 c轴的一维传导通道的存在。由Lotsch等[64]最近发现的 Li11Si2PS12、Li10SnP2S12与Li10GeP2S12具有相似的晶体结构。图9为不同电解质的离子电导率随温度的变化规律,可以看出,和其它固体电解质材料相比,Li10GeP2S12三元硫化物固体电解质在低于 100℃时是迄今发现的室温锂离子电导率最高的材料。

图8 (a)参与离子传导的框架结构和锂离子;(b)Li10GeP2S12框架结构;(c)Li+迁移通道[63]Fig.8 (a) The framework structure and lithium ions that participate in ionic conduction;(b) Framework structure of Li10GeP2S12;(c) Conduction pathways of lithium ions[63]
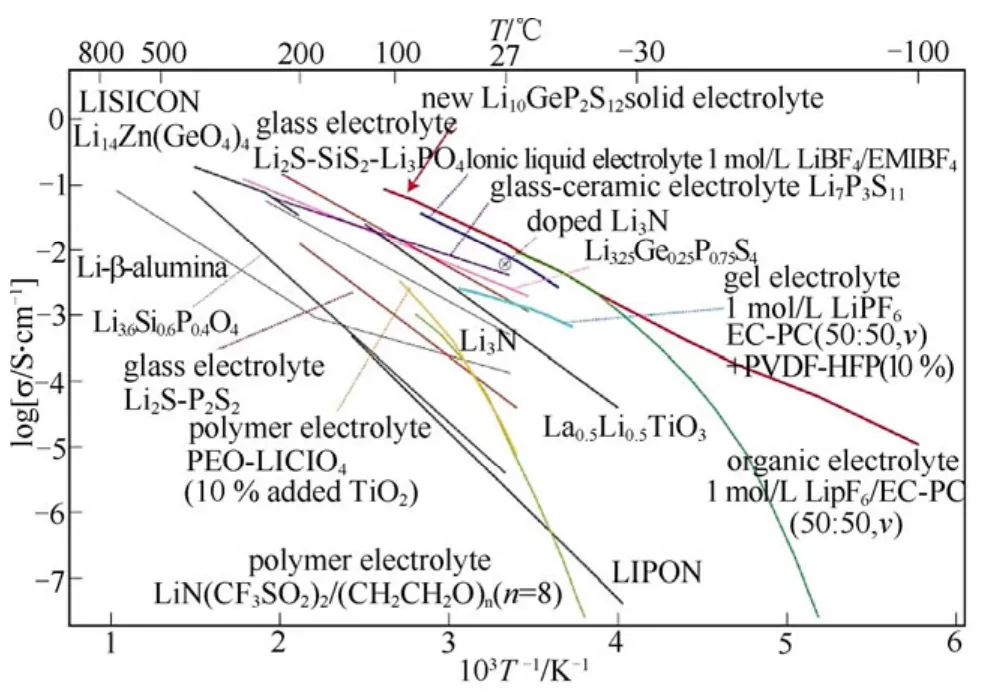
图9 不同电解质的离子电导率图[63] Fig.9 Ion conductivity for different type of electrolytes[63]
3.2 Li2S-P2S5二元硫化物锂离子固体电解质
3.2.1 制备方法
硫化物电解质的制备方法主要有三种:一种是熔融法,即将原料按一定化学计量比混合后得到初料,初料经高温熔化后转变为熔融态,熔融态初料淬火即可得玻璃态硫化物固体电解质,玻璃态硫化物固体电解质经过析晶可进一步得到玻璃陶瓷态硫化物固体电解质;另一种制备方法是高能球磨法,即先将原料混合,高能球磨一定时间后得玻璃态硫化物固体电解质,将玻璃态硫化物固体电解质析晶可得玻璃陶瓷态硫化物固体电解质;第三种制备方法是液相法,即将原料按一定化学计量比分别加入到有机溶剂中,在适宜的反应温度下搅拌一定时间,反应结束后通过离心或旋蒸将液态溶剂去除,在一定的温度下干燥即可得到非晶硫化物固体电解质材料,进一步析晶即可得到陶瓷类硫化物固体电解质材料。三种制备方法的优缺点见表3。

表3 硫化物固体电解质三种制备方法的优缺点 Table 3 Advantages and disadvantages of the three preparation methods of sulphide solid electrolytes
Minami等[62]采用熔融法将 70%Li2S-30%P2S5初料分别在750℃、850℃温度下烧结20 h和950 ℃温度下烧结30 min并淬火,获得了3种不同热处理温度条件下的70%Li2S-30%P2S5玻璃态电解质;同时采用高能球磨法室温球磨 20 h制备相同组分的70%Li2S-30%P2S5玻璃态电解质材料,然后将在上述4种条件下制备的70%Li2S-30%P2S5玻璃在相同条件下析晶,得到70%Li2S-30%P2S5玻璃陶瓷。结果表明,采用高能球磨法和在750 ℃热处理20 h时所得材料的组成一致,其离子电导率均可达2.1×10-3S/cm,高于其它条件下制备的70%Li2S-30%P2S5玻璃陶瓷电解质。这表明熔融法和高能球磨法是制备同种材料的两种不同方法,但高能球磨法由于可在室温进行、安全系数更高等特点,是制备硫化物固体电解质的常规方法。值得一提的是,Liu等[65]最近采用液相化学法成功制备了含有纳米级 β-Li3PS4快离子导体的 75%Li2S-25%P2S5硫化物固体电解质。但室温离子电导率较低,约为1×10-4S/cm,电化学窗口约为5.0 V vs Li+/Li。虽然制备的硫化物电解质离子电导率较低,但这种新的合成方法为今后制备硫化物固体电解质开辟了一条新的道路。
3.2.2 影响性能的主要因素
(1)高能球磨工艺和析晶条件 高能球磨工艺和析晶条件是影响硫化物固体电解质性能的两个重要因素。常用的高能球磨工艺有两种:一种是采用10个直径为10 mm氧化锆球和45 mL氧化锆球磨罐,370 r/min 转速下球磨 20 h[60, 66-68]。另一种是采用160个直径为4 mm的氧化锆球和45 mL球磨罐,510 r/min转速下球磨10 h以上[69-71]。此外,Ujiie等[72]还采用 500个直径为 4 mm 的氧化锆球和 45 mL球磨罐,500 r/min转速下球磨10 h后成功制备出性能较好的硫化物电解质材料。球料比、球磨珠和球磨罐的材质亦对电解质材料有较大的影响。高能球磨法一般为干磨,为了避免引入杂质,对球磨珠和球磨罐的材质有较高要求:耐磨,在高速球磨碰撞中不易碎,不掉粉。
析晶条件主要包括析晶温度和析晶时间,相关研究表明[60],析晶温度过高或过低、析晶时间过长或过短均会降低电解质的电化学性能和化学性能。这是因为析晶温度过低,Li2S晶体析出;析晶温度过高,电解质材料发生分解,降低材料的离子电导率。而析晶时间过长和过短,会减少 Li+离子在电解质中的传输通道,导致材料的锂离子电导率降低。因此,析晶过程中须严格控制析晶温度和析晶时间。
(2)硫化物固体电解质的物相结构组成和结构是影响硫化物固体电解质性能最重要的因素。与晶态材料相比,由于玻璃长程无序、短程有序和各向同性等特点,将硫化物电解质制成玻璃态,可进一步扩大电解质中锂离子的传输通道,获得具有较高离子电导率的电解质材料。一般而言,玻璃态硫化物电解质的室温锂离子电导率可达 10-4S/cm,然而,将玻璃态硫化物电解质材料析晶所得的玻璃陶瓷态电解质材料的室温锂离子电导率更高,可达10-3S/cm。这是因为在析晶过程中,Li2S-P2S5无定形粉末发生软化,降低了Li2S-P2S5电解质中的晶界电阻;此外,部分晶态超离子导体的析出也有助于提高材料的晶粒锂离子电导率。
Hayashi等[73]对比了 70%Li2S-30%P2S5玻璃和70%Li2S-30%P2S5玻璃陶瓷的电化学性能。结果显示,70%Li2S-30%P2S5玻璃和 70%Li2S-30%P2S5玻璃陶瓷在高温时(约300 ℃)的离子电导率相近,但在室温至200℃的温度范围内,70%Li2S-30%P2S5玻璃的离子电导率低于70%Li2S-30%P2S5玻璃陶瓷的离子电导率。
(3)硫化物固体电解质的组分构成 物相对材料性能的影响直接体现在表观形貌上,原料组分则是从内在影响材料的性能。改变Li、P和S等之间的比例,所得材料的组成不同。当Li2S∶P2S5为7∶3、2∶1或者 3∶1(摩尔比)时,根据 Li∶P∶S三者的化学计量比,分别优先形成Li7P3S11、Li4P2S7和Li3PS4三种快离子导体晶相。但实际反应过程中,即使严格按照化学计量比,由于原料不能充分均匀混合,在局部区域Li2S和P2S5的比例发生改变,生成少量杂相。因此,Li2S和P2S5的摩尔比例不同,各项制备参数也将不同,材料的电学性能和电化学性能亦不同。
Hayashi等[67]采用高能球磨法研究了不同组分x%Li2S-(100-x) %P2S5(x=50、60、70、75、80、87.5)玻璃的性能。结果表明,当 x≥80%时,所需球磨时间大于35 h;而当x≤75时,球磨时间仅需20 h。对于上述多种玻璃态Li2S-P2S5电解质,其离子电导率 σ 的大小顺序依次为 σ(x=75)> σ(x=80)> σ(x=70)> σ(x=87.5),其中 75%Li2S-25%P2S5玻璃的离子电导率为 2×10-4S/cm。此外,80%Li2S-20%P2S5玻璃陶瓷[74]的室温离子电导率可达9×10-4S/cm以上,而70%Li2S-30%P2S5玻璃陶瓷[75]的离子电导率高达3.2×10-3S/cm。目前,在仅由Li2S和P2S5二者组成的纯组分硫化物电解质材料中,70%Li2S-30%P2S5玻璃陶瓷的离子电导率最高。虽然 70%Li2S- 30%P2S5玻璃陶瓷具有较高的离子电导率,但和诸多硫化物电解质相似,其空气稳定性不佳,短时间暴露于空气中即会反生吸潮反应[76],如下式所示:
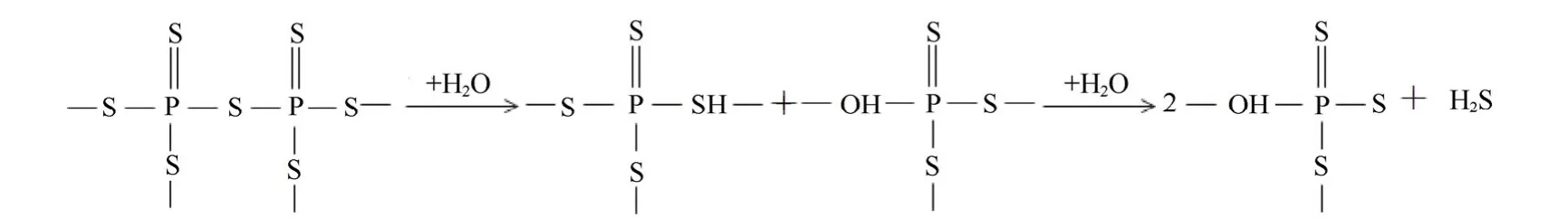
这是因为硫化物电解质中一般都含有桥接硫,桥接硫遇水极易发生吸潮反应。研究表明,虽然在诸多二元纯组分硫化物固体电解质中75%Li2S-25%P2S5玻璃的离子电导率相对偏低,但其空气稳定性最佳。该电解质暴露于空气中7 h,其锂离子电导率保持率达到95%以上,其主要原因是该组分硫化物固体电解质中桥接硫的含量非常低。因此,控制桥接硫含量对提高硫化物电解质的空气稳定性具有非常重要的作用,控制组成比例是提高硫化物电解质的电化学性能和化学稳定性的一个重要途径。
本文作者所在课题组通过系统研究不同组分(100-x)%Li2S-x%P2S5(摩尔分数)(简称 LPS)玻璃陶瓷的离子电导率随x值的变化规律,选取x=20、25和30,。当x=20时,即LPS@8-2 G-C,其晶相是Li8P2S9,该结构由Li3PS4(2个)和Li2S(1个)两种晶相固溶形成;当x=25时,即LPS@3-1 G-C,其晶相是Li3PS4,属于β-Li3PO4结构;当x=30时,即 LPS@7-3 G-C,其晶相是 Li7P3S11,该结构由Li4P2S7和Li3PS4两种晶相固溶形成。图10显示了LPS@8-2 G-C、LPS@7-3 G-C 和 LPS@3-1 G-C 三种组分材料玻璃陶瓷的室温阻抗图谱,为了进行比较,列出了LPS@7-3 G玻璃材料的室温阻抗图谱。通过计算获得各材料的室温锂离子电导率,如表 4所示,对LPS@7-3组分材料而言,玻璃陶瓷化以后,其离子电导率提升了近一个数量级,这也证实了玻璃陶瓷材料具有更好的离子导电性能。对不同组分的玻璃陶瓷材料,LPS@7-3 G-C的室温离子电导率最高,达到 9.31×10-4S/cm;LPS@3-1 G-C 组分的最低,为 4.02×10-4S/cm。离子电导率跟组分密切相关的原因主要是材料导电主晶相结构发生了变化。
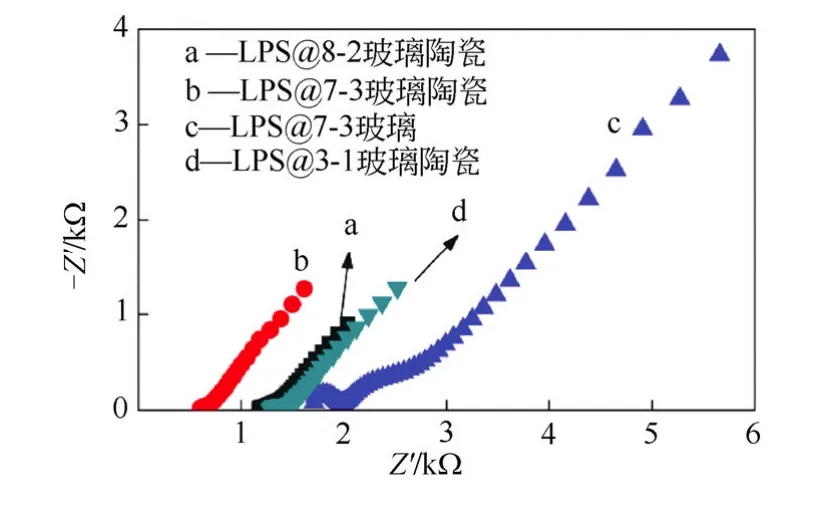
图10 LPS固体电解质材料的室温阻抗谱 Fig.10 The EIS plots of different sulfide solid electrolytes

表4 LPS固体电解质材料的导电相、室温离子电导率和激活能 Table 4 Conductive phase, conductivities and activation energy of LPS solid electrolytes
3.2.3 改性研究
硫化物固体电解质虽然具有较高的锂离子电导率和优良的电化学性能,但其空气稳定性不佳;而空气稳定性相对较好的硫化物固体电解质的锂离子电导率却相对偏低。为了获得即具有较好电学、电化学性能,又具有较好化学稳定性的硫化物固体电解质,研究者们对硫化物类固体电解质材料进行了改性实验,获得了较好的研究成果。
Ohtomo等[77]用 P2O5取代部分 P2S5,制备了Li2S-P2S5-P2O5玻璃陶瓷电解质。结果表明,虽然添加P2O5后材料的离子电导率有所降低,但其电化学窗口更宽,可达 5 V vs.Li+/Li以上。Minami等[78]将P2S3取代70%Li2S-30%P2S5中的部分P2S5,P2S3的加入有效抑制了Li4S2P6慢离子导体的产生,提高了材料的锂离子电导率。测试结果表明,所制备的70%Li2S-29%P2S5-1%P2S3的室温离子电导率高达3.9×10-3S/cm 。Ujiie等[72]则研究了在70%Li2S-30%P2S5中掺杂LiI的可行性,结果表明,70%Li2S-30%P2S5的锂离子电导率在引入 LiI后迅速降低,LiI的最佳摻入量为20%,此时其室温锂离子电导率仅为5.6×10-4S/cm,但其电化学窗口高达10 V vs.Li+/Li以上。此外,在Li2S-P2S5电解质中掺杂或复合部分 SiS2[79]、Sb2S3[80]或 Li2O[81]等也能有效改善电解质的电化学性能。Ohtomo等[82]在75%Li2S-25%P2S5电解质掺杂部分FeS和CuO,结果表明,掺杂FeS或CuO后电解质的空气稳定性得到较好的提高。此外,Ohtomo等[83]还采用“两步法”在 Li2S-P2S5电解质中掺杂和复合了部分 Li2O,结果表明材料的空气稳定性得到了极大的提升。将所制备的 xLi2O-(100-x)(0.7Li2S-0.3P2S5)暴露于空气中5min,未检测到H2S的产生,材料的电化学性能未发生明显变化。这表明,对硫化物电解质进行掺杂改性或者复合改性,不仅有助于改善材料的电化学性能,还有助于提高材料的化学稳定性。
本文作者所在课题组的研究表明,LPS@7-3玻璃陶瓷的晶相是Li7P3S11,离子电导率可以达到9.31×10-4S/cm。但是,其离子电导率有进一步提升空间,同时,为了满足实用的要求,还需要在电化学性能方面综合考量,主要包括以金属锂为参比电极下的氧化-还原电位,即电化学窗口以及金属锂为电极时的化学稳定性。因此,首先围绕LPS@7-3进行掺杂改性,以获得综合性能优异的固体电解质材料进行实用。在LPS的玻璃网络中掺杂引入氧原子,有望提高材料的热稳定性和离子电导率。其原因就在于氧原子的引入在玻璃体网络中是以共价键链接的,P—O键相比于P—S键具有更强的结合,从而导致P—O键的结构单元对锂离子具有相对较弱的作用力,这将有利于锂离子的快速迁移。所以,首先采用微量 Li3PO4掺杂 70%Li2S-30%P2S5以形成70%Li2S-(30-x)%P2S5-x%Li3PO4(x=0~5)体系材料。
图11为不同掺杂量 70%Li2S-(30-x)%P2S5- x%Li3PO4玻璃陶瓷的室温锂离子电导率与组分的关系曲线。从图中可看出,随着掺杂量的增加,材料的总电导率先增加后减少。当x=1时,玻璃陶瓷材料的总离子电导率最高,为 σt= 1.68×10-3S/cm,掺杂后,离子电导率提升了超过 80%。图 12为70%Li2S-29%P2S5-1%Li3PO4玻璃陶瓷的 CV曲线图。从图中可以看出,该玻璃陶瓷锂离子导体具有宽的电化学窗口,其电化学窗口可达10 V vs Li+/Li。

图11 70%Li2S-(30-x)%P2S5-x%Li3PO4玻璃陶瓷的电导率-组分关系曲线 Fig.11 The composition dependence of ionic conductivities of 70%Li2S-(30-x)%P2S5-x%Li3PO4

图12 70%Li2S-29%P2S5-1%Li3PO4玻璃陶瓷的CV曲线 Fig.12 The CV curve of 70%Li2S-29%P2S5-1%Li3PO4glass ceramics
本文作者所在课题组对不同掺杂量 70%Li2S- (30-x)%P2S5-x%P2O5(x=0~5)玻璃陶瓷也进行系统研究,当x=1时,玻璃陶瓷材料的总离子电导率最高,σt= 1.87×10-3S/cm,其电化学窗口也可达 10 V vs Li+/Li。随后,他们研究了 70%Li2S-30%P2S5、70%Li2S-29%P2S5-1%Li3PO4和 70%Li2S-29%P2S5- 1%P2O53种组分玻璃陶瓷对锂阻抗随时间变化关系曲线,如图13所示,可以看出,70%Li2S-30%P2S5玻璃陶瓷接触金属锂的长期稳定性较差,其阻抗值增大显著;70%Li2S-29%P2S5-1%Li3PO4玻璃陶瓷接触金属锂的长期稳定性相对未掺杂材料有明显改善,但还是会有所增大,尤其是在开始阶段; 70%Li2S-29%P2S5-1%P2O5玻璃陶瓷接触金属锂的长期稳定性在3种材料中是最好的,该材料能够与金属锂保持良好的兼容性。
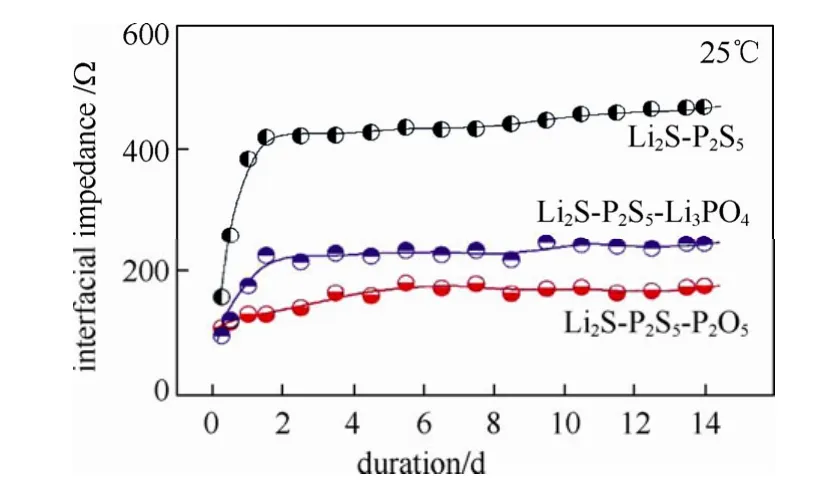
图13 Li/LPS基固体电解质/Li的界面阻抗变化曲线图 Fig.13 The interface impedance of Li/LPS-based solid electrolytes/Li
3.3 Li2S-P2S5-GeS2三元硫化物固体电解质
虽然研究者们对 Li2S-P2S5硫化物体系进行了诸多改性实验,但材料的电化学性能一直未得到明显提高,电解质的室温离子电导率一般低于3×10-3S/cm。为了寻找更加有效的改性物质,提高硫化物电解质的化学稳定性和电化学性能,研究人员尝试在 Li2S-P2S5体系中引入 GeS2。Kamaya等[63]将 Li2S、P2S5和GeS2振动磨混料30 min后获得初料,初料经压片、550 ℃热处理 8 h、随炉冷却后制得Li10GeP2S12硫化物电解质。该材料的室温离子电导率高达12 mS/cm(可媲美液态电解质),电化学窗口为5 V vs.Li+/Li。该电解质与LiCoO2正极材料和In负极材料一起组装成的全固态锂电池,在 14 mA/g电流密度下,充放电8次后的容量仍大于120 mA·h/g,第二圈充放电的库仑效率约为 100%。最近出现了研究Si基和Sn基的LGPS型固体电解质的热潮。Ceder等[84]利用第一性原理研究Li10±1MP2X12(M=Ge,Si,Sn,Al 或 P,X=O,S或 Se)超离子导体发现,相对于 Li10GeP2S12硫化物电解质,Si、Sn等价离子取代Ge后,具有类似的相稳定性、电化学稳定性和锂离子电导率;而异价离子Al和P取代Ge后,离子电导率减小。阴离子取代则影响更为显著,当O取代S后,离子电导率显著降低,而当Se取代S后,虽然锂离子电导率得到提高,却造成了电化学稳定性变差,并认为骨架结构中S2-大小最适合Li离子迁移。Dehnen和Roling等[85]则用实验制备了Li10SnP2S12固体电解质材料,其室温总离子电导率为 4 mS/cm,相比于Li10GeP2S12硫化物电解质的12 mS/cm低,但却大幅度降低了原料的成本。
Lotsch等[86]则最近首次发现了 Li7GePS8硫化物电解质,并同时将其与Li10GeP2S12硫化物电解质进行对比,发现合成的 Li7GePS8和 Li10GeP2S12硫化物电解质室温离子电导率分别为 7 mS/cm 和 9 mS/cm,这与其锂离子在体相晶格中的各向同性的跳跃机制有关,二者的激活能均约为0.22 eV。随后,Lotsch等[64]又率先报道了新的超离子导体Li11Si2PS12,发现其室温离子扩散系数比现有最好的Li10GeP2S12硫化物固体电解质还要更高,并讨论了LMePS(Me=Si、Ge、Sn等)家族成员的结构和性能。LMePS家族电解质的通式为 L11-xMe2-xP1+xS12(Me=Si、Ge、Sn 等),Li10GeP2S12、Li7GePS8、Li10SnP2S12。四方 Li10SnP2S12是通过对 Sn、P、S元素和Li2S在653 K的温度下加热10 h,然后在723 K下烧结2天制备所得的。离子半径较大的四价Sn位于六配位而非四配位间隙,这体现在当原料严格按照化学计量比使用时,SnS6八面体结构单元以共棱形式构成杂相层(如 LiSnS3)。而当 Li2S过量10%~20%后将会彻底阻碍这些杂相层的形成。另一方面,Li11Si2PS12是利用高压合成法制备的,若按照Li10SiP2S12计量比称量,采用传统固相烧结的方式烧结,在 573~1023 K的范围内烧结后的样品主晶相为正交畸变后的Li11Si2PS12材料,而这种材料的离子传导特性大大降低。
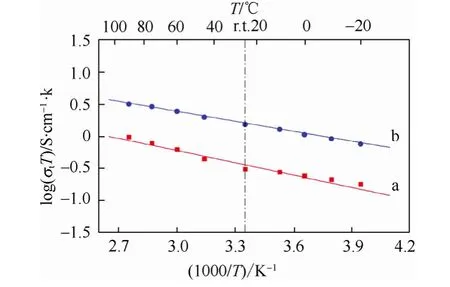
图14 二元(曲线a)和三元(曲线b)硫化物固体电解质材料的离子电导率随温度的变化 Fig.14 The temperature dependence of ionic conductivities of (a) Li2S-P2S5and (b) Li2S-P2S5-GeS2
本文作者所在课题组对 Li10GeP2S12硫化物固体电解质材料也进行了研究,并将其离子电导率随温度的变化和二元硫化物固体电解质进行了对比,如图14所示。作者制备的第一代三元硫化物固体电解质的室温离子电导率超过6 mS/cm,相对于二元的0.93 mS/cm有了很大的提升,经过计算二者的激活能分别为0.20 eV和0.22 eV。将此两种硫化物固体电解质材料装配于 LiCoO2基全固态锂电池中,通过在室温下0.1C倍率时充放电性能的对比,可以清楚看出,与三元电解质相比,使用二元电解质时LiCoO2基正极材料充放电时存在较大的极化现象,而且首次放电比容量也明显偏低,约为90 mA·h/g,而三元硫化物基全固态锂电池的首次放电比容量超过120 mA·h/g,这是由于二元电解质室温锂离子电导率远低于三元电解质所致。

图15 基于LiCoO2正极的全固态锂电池的首次充放电曲线 Fig.15 Initial charge-discharge curves of all-solid-state lithium cells using LiCoO2as a cathode material
4 结 语
采用无机固体电解质可以从根本上解决锂离子电池的安全问题,而且避免了电解质与电极的副反应,是高安全性锂离子电池未来发展的必然趋势。但是无机固体电解质材料也存在着室温离子电导率和高稳定性的矛盾,即高离子电导率的硫化物固体电解质材料空气稳定性不足,而稳定性良好的氧 化物固体电解质材料其室温离子电导率又存在着 瓶颈。
目前,NASICON结构型和石榴石结构型氧化物固体电解质材料、硫化物固体电解质材料是目前最有可能应用到全固态锂电池中的固体电解质材料。NASICON结构型和石榴石结构型氧化物固体电解质材料具有优异的空气稳定性,室温离子电导率可达10-3S/cm数量级;硫化物固体电解质由于具有较好的电化学性能、化学性能和较宽的电化学窗口,特别是其室温锂离子电导率可达10-2S/cm数量级,是目前发展最快的固体电解质材料之一,但其空气稳定性有待提高。
另一方面,对全固态锂电池用固体电解质材料的研究不能仅仅局限于其离子电导率和电化学窗口等方面,固体电解质材料与正负极材料间的电化学和化学稳定兼容性也是构造全固态锂电池所必须要考虑的关键因素。只有充分认识到固体电解质材料在全固态锂电池中的核心作用和应用过程中的关键科学问题,才可能研究开发出适用于大容量全固态锂电池的锂离子固体电解质材料。
[1] Peng Jiayue(彭佳悦),Zu Chenxi(祖晨曦),Li Hong(李泓).Fundamental scientific aspects of lithium batteries(I)—Thermodynamic calculations of theoretical energy densities of chemical energy storage systems[J].Energy Storage Science and Technology(储能科学与技术),2013,2(1):55-62.
[2] Xu Xiaoxiong(许晓雄),Qiu Zhijun(邱志军),Guan Yibiao(官亦标),Huang Zhen(黄祯),Jin Yi(金翼).All-solid-state lithium-ion batteries:State-of-the-art development and perspective[J].Energy Storage Science and Technology(储能科学与技术),2013,2(4):331-341.
[3] Lin Zuxiang(林祖纕),Guo Zhukun(郭祝昆).Fast Ionic Conductor(快离子导体)[M].Shanghai:Shanghai Science and Technology Press(上海科学技术出版社),1983.
[4] Dong Xiaochen(董晓臣),Wang Li(王立).Compositions,structures and properties of polymer electrolytes for lithium ion battery[J].Progress in Chemistry(化学进展),2005,17(2):248-253.
[5] Xu X,Wen Z,Wu J,Yang X.Preparation and electrical properties of NASICON-type structured Li1.4Al0.4Ti1.6(PO4)3glass-ceramics by the citric acid-assisted sol-gel method[J].Solid State Ionics,2007,178(1-2):29-34.
[6] Xu X,Wen Z,Yang X,Zhang J,Gu Z.High lithium ion conductivity glass-ceramics in Li2O-Al2O3-TiO2-P2O5from nanoscaled glassy powders by mechanical milling[J].Solid State Ionics,2006,177(26-32):2611-2615.
[7] Liu Wenyuan(刘文元),Fu Zhengwen(傅正文),Qin Qizong(秦启宗).Studies on lithium phosphorous oxynitride electrolyte thin films and a new all-solid-state thin film lithium battery[J].Acta Chimica Sinica(化学学报),2004,62(22):2223-2227.
[8] Hagman L,Kierkegaard P.The crystal structure of NaMe2IV(PO4)3; MeIV= Ge,Ti,Zr [J].Acta Chemica Scandinavica,1968,22(6):1822-1832.
[9] Goodenough J B,Hong H Y P,Kafalas J A.Fast Na+-ion transport in skeleton structures[J].Materials Research Bulletin,1976,11(2):203-220.
[10] Anantharamulu N,Rao K K,Rambabu G,Kumar B V,Radha V,Vithal M.A wide-ranging review on NASICON type materials[J].Journal of Materials Science,2011,46(9):2821-2837.
[11] Thangadurai V,Weppner W J F.Recent progress in solid oxide and lithium ion conducting electrolytes research[J].Ionics,2006,12(1):81-92.
[12] Fergus J W.Ceramic and polymeric solid electrolytes for lithium-ion batteries[J].Journal of Power Sources,2010,195(15):4554-4569.
[13] Zheng Yuelei(郑玥雷),Chen Renjie(陈人杰),Wu Feng(吴锋),Li Li(李丽).Progress of research on the conductive mechanism of the glassy electrolytes in lithium ion batteries[J].Journal of Inorganic Materials(无机材料学报),2013,28(11):1172-1180.
[14] Zheng Hao(郑浩),Gao Jian(高健),Wang Shaofei(王少飞),Li Hong( 李泓) .Fundamental scientific aspects of lithium batteries(VI)—Ionic transport in solids[J].Energy Storage Science and Technology(储能科学与技术),2013,2(6):620-635.
[15] Aono H,Sugimoto E,Sadaoka Y,Imanaka N,Adachi G Y.Ionic conductivity of solid electrolytes based on lithium titanium phosphate[J].Journal of the Electrochemical Society,1990,137(4):1023-1027.
[16] Chowdari B V R,Rao G V S,Lee G Y H.XPS and ionic conductivity studies on Li2O-Al2O3(TiO2or GeO2)-P2O5glass-ceramics[J].Solid State Ionics,2000,136:1067-1075.
[17] Leo C J,Chowdari B V R,Rao G V S,Souquet J L.Lithium conducting glass ceramic with NASICON structure[J].Materials Research Bulletin,2002,37(8):1419-1430.
[18] Zheng Honghe(郑洪河),Qu Qunting(曲群婷),Liu Yunwei(刘云伟),Xu Zhongyu(徐仲榆).Research progress of inorganic solid electrolyte materials for lithium and lithium ion batteriesⅠ Lithium ceramic solid electrolytes[J].Chinese Journal of Power Sources(电源技术),2007,131(5):349-353.
[19] Zheng Honghe(郑洪河),Qu Qunting(曲群婷),Shi Jing(石静),Xu Zhongyu(徐仲榆).Research progress of inorganic solid electrolyte materials for lithium and lithium ion batteries Ⅱ Glassy-state lithium inorganic solid electrolytes[J].Chinese Journal of Power Sources(电源技术),2007,31(12):1015-1020.
[20] Zhu Yongming(朱永明),Ren Xuefeng(任雪峰),Li Ning(李宁).Progress of inorganic solid state lithium ion electrolyte[J].Chemistry Online(化学通报),2010(12):1073-1079.
[21] Kotobuki M,Hoshina K,Isshiki Y,Kanamura K.Preparation of Li1.5Al0.5Ge1.5(PO4)3solid electrolyte by sol-gel method[J].Phosphorus Research Bulletin,2011,25:61-63.
[22] Duluard S,Paillassa A,Puech L,Vinatier P,Turq V,Rozier P,Lenormand P,Taberna P L,Simon P,Ansart F.Lithium conducting solid electrolyte Li1.3Al0.3Ti1.7(PO4)3obtained via solution chemistry[J].Journal of the European Ceramic Society,2013,33(6):1145-1153.
[23] Kotobuki M,Koishi M,Kato Y.Preparation of Li1.5Al0.5Ti1.5(PO4)3solid electrolyte via a co-precipitation method[J].Ionics,2013,19(12):1945-1948.
[24] Xu X X,Wen Z Y,Yang X L,Chen L D.Dense nanostructured solid electrolyte with high Li-ion conductivity by spark plasma sintering technique[J].Materials Research Bulletin,2008,43(8-9):2334-2341.
[25] Morimoto H,Awano H,Terashima J,Shindo Y,Nakanishi S,Ito N,Ishikawa K,Tobishima S.Preparation of lithium ion conducting solid electrolyte of NASICON-type Li1+xAlxTi2-x(PO4)3(x=0.3) obtained by using the mechanochemical method and its application as surface modification materials of LiCoO2cathode for lithium cell[J].Journal of Power Sources,2013,240:636-643.
[26] Xiao Zhuobing(肖卓炳),Chen Shang(陈上),Guo Manman(郭满满).Influence of Li3PO4addition on properties of lithium ion-conductive electrolyte Li1.3Al0.3Ti1.7(PO4)3[J].Transactions of Nonferrous Metals Society of China,2011,21(11):2454-2458.
[27] Aono H,Sugimoto E,Sadaoka Y,Adachi G.The electrical properties of ceramic electrolytes for LiMxTi2-x(PO4)3+y-Li2O,M=Ge,Sn,Hf,and Zr systems[J].Journal of the Electrochemical Society,1993,140:1827-1832.
[28] Kotobuki M,Koishi M.Preparation of Li1.5Al0.5Ti1.5(PO4)3solid electrolyte via a sol-gel route using various Al sources[J].Ceramics International,2013,39(4):4645-4649.
[29] Zhang P,Matsui M,Hirano A,Takeda Y,Yamamoto O,Imanishi N.Water-stable lithium ion conducting solid electrolyte of the Li1.4Al0.4Ti1.6-xGex(PO4)3system(x=0~1.0)with NASICON-type structure[J].Solid State Ionics,2013,253:175-180.
[30] Johnson P,Sammes N,Imanishi N,Takeda Y,Yamamoto O.Effect of microstructure on the conductivity of a NASICON-type lithium ion conductor[J].Solid State Ionics,2011,192(1):326-329.
[31] Kumar B,Thokchom J S.Space charge signature and its effects on ionic transport in heterogeneous solids[J].Journal of the American Ceramic Society,2007,90(10):3323-3325.
[32] Xu X,Wen Z,Wu X,Yang X,Gu Z.Lithium ion-conducting glass-ceramics of Li1.5Al0.5Ge1.5(PO4)3-xLi2O(x=0~0.20)with good electrical and electrochemical properties[J].Journal of the American Ceramic Society,2007,90(9):2802-2806.
[33] Fu J.Fast Li+ion conducting glass-ceramics in the system Li2O-Al2O3-GeO2-P2O5[J].Solid State Ionics,1997,104(3):191-194.
[34] He K,Wang Y H,Zu C K,Zhao H F,Liu Y H,Chen J,Han B,Ma J R.Influence of Al2O3additions on crystallization mechanism and conductivity of Li2O-Ge2O-P2O5glass-ceramics[J].Physica B,2011,406(20):3947-3950.
[35] Fu J.Effects of M3+ions on the conductivity of glasses and glass-ceramics in the system Li2O-M2O3-GeO2-P2O5(M= Al, Ca, Y, Dy, Gd, and La)[J].Journal of the American Ceramic Society,2000,83(4):1004-1006.
[36] Katoh T,,Inda Y,Baba M,Ye R.Lithium-ion conductive glass-ceramics with composition ratio control and their electrochemical characteristics[J].Journal of the Ceramic Society of Japan,2010,118(1384):1159-1162.
[37] Jadhav H S,Cho M S,Kalubarme R S,Lee J S,Jung K N,Shin K H,Park C J.Influence of B2O3addition on the ionic conductivity of Li1.5Al0.5Ge1.5(PO4)3glass ceramics[J].Journal of Power Sources,2013,241:502-508.
[38] Cruz A M,Ferreira E B,Rodrigues A C M.Controlled crystallization and ionic conductivity of a nanostructured LiAlGePO4glass-ceramic[J].Journal of Non-Crystalline Solids,2009,355(45-47):2295-2301.
[39] Thokchom J S,Gupta N,Kumar B.Superionic conductivity in a lithium aluminum germanium phosphate glass-ceramic[J].Journal of the Electrochemical Society,2008,155(12):A915-A920.
[40] Kasper H M.A new series of rare earth garnets Ln3+3M2Li+3O12(M=TE, W)[J].Inorganic Chemistry,1969,8(4):1000-1002.
[41] Mazza D.Remarks on a ternary phase in the La2O3-Nb2O5-LI2O,La2O3-Ta2O5-Li2O system[J].Materials Letters,1988,7(5-6):205-207.
[42] Hyooma H,Hayashi K.Crystal-structures of La3Li5Nb2O12,La3Li5Ta2O12[J].Materials Research Bulletin,1988,23(10):1399-1407.
[43] Cussen E J.The structure of lithium garnets:Cation disorder and clustering in a new family of fast Li+conductors[J].Chemical Communications,2006(4):412-413.
[44] Thangadurai V,Kaack H,Weppner W J F.Novel fast lithium ion conduction in garnet-type Li5La3M2O12(M = Nb, Ta)[J].Journal of the American Ceramic Society,2003,86(3):437-440.
[45] Thangadurai V,Weppner W J F.Li6ALa2Ta2O12(A=Sr, Ba):Novel garnet-like oxides for fast lithium ion conduction[J].Advanced Functional Materials,2005,15(1):107-112.
[46] Thangadurai V,Weppner W J F.Li6ALa2Nb2O12(A = Ca,Sr,Ba):A new class of fast lithium ion conductors with garnet-like structure[J].Journal of the American Ceramic Society,2005,88(2):411-418.
[47] Murugan R,Thangadurai V,Weppner W J F.Fast lithium ion conduction in garnet-type Li7La3Zr2O12[J].Angewandte Chemie:International Edition,2007,46(41):7778-7781.
[48] Thangadurai V,Weppner W J F.Effect of sintering on the ionic conductivity of garnet-related structure Li5La3Nb2O12and In- and K-doped Li5La3Nb2O12[J].Journal of Solid State Chemistry,2006,179(4):974-984.
[49] Awaka J,Kijima N,Hayakawa H,Akimoto J.Synthesis and structure analysis of tetragonal Li7La3Zr2O12with the garnet-related type structure[J].Journal of Solid State Chemistry,2009,182(8):2046-2052.
[50] Geiger C A,Alekseev E,Lazic B,Fisch M,Armbruster T,Langner R,Fechtelkord M,Kim N,Pettke T,Weppner W J F.Crystal chemistry and stability of "Li7La3Zr2O12" Garnet:A fast lithium-ion conductor[J].Inorganic Chemistry,2011,50(3):1089-1097.
[51] Adams S ,Rao R P.Ion transport and phase transition in Li7-xLa3(Zr2-xMx)O12(M = Ta5+,Nb5+,x=0,0.25)[J].Journal of Materials Chemistry,2012,22(4):1426-1434.
[52] Murugan R,Ramakumar S,Janani N.High conductive yttrium doped Li7La3Zr2O12cubic lithium garnet[J]. Electrochemistry Communications,2011,13(12):1373-1375.
[53] Ohta S,Kobayashi T,Asaoka T.High lithium ionic conductivity in the garnet-type oxide Li7-xLa3(Zr2-x, Nbx)O12(x=0~2)[J].Journal of Power Sources,2011,196(6):3342-3345.
[54] Allen J L,Wolfenstine J,Rangasamy E,Sakamoto J.Effect of substitution(Ta,Al,Ga) on the conductivity of Li7La3Zr2O12[J].Journal of Power Sources,2012,206:315-319.
[55] Li Y,Han J T,Wang C A,Xie H,Goodenough J B.Optimizing Li+conductivity in a garnet framework[J].Journal of Materials Chemistry,2012,22(30):15357-15361.
[56] Dhivya L,Janani N,Palanivel B,Murugan R.Li+transport properties of W substituted Li7La3Zr2O12cubic lithium garnets[J].Aip Advances,2013,3(8):082115.
[57] Deviannapoorani C,Dhivya L,Ramakumar S,Murugan R.Lithium ion transport properties of high conductive tellurium substituted Li7La3Zr2O12cubic lithium garnets[J].Journal of Power Sources,2013,240:18-25.
[58] Wu Yuping(吴宇平),Yuan Xiangyun(袁祥云),Dong Chao(董超),Duan Jiyuan(段冀渊).Lithium Battery—Application and Practice(锂离子电池——应用与实践)[M].Beijing:Chemical Industry Press(化学工业出版社),2011:249-250.
[59] Kanno R,Maruyama M.Lithium ionic conductor thio-LISICON—The Li2S-GeS2-P2S5system[J].Journal of the Electrochemical Society,2001,148(7):A742-A746.
[60] Hayashi A,Hama S,Minami T,Tatsumisago M.Formation of superionic crystals from mechanically milled Li2S-P2S5glasses[J].Electrochemistry Communications,2003,5(2):111-114.
[61] Mizuno F,Hayashi A,Tadanaga K,Tatsumisago M.New,highly ion-conductive crystals precipitated from Li2S-P2S5glasses[J].Advanced Materials,2005,17(7):918-921.
[62] Minami K,Mizuno F,Hayashi A,Tatsumisago M.Lithium ion conductivity of the Li2S-P2S5glass-based electrolytes prepared by the melt quenching method[J].Solid State Ion.,2007,178(11-12):837-841.
[63] Kamaya N,Homma K,Yamakawa Y,Hirayama M,Kanno R,Yonemura M,Kamiyama T,Kato Y,Hama S,Kawamoto K,Mitsui A.A lithium superionic conductor[J].Nature Materials,2011,10(9):682-686.
[64] Kuhn A,Gerbig O,Zhu C,Falkenberg F,Maier J,Lotsch B V.A new ultrafast superionic Li-conductor:Ion dynamics in Li11Si2PS12and comparison with other tetragonal LGPS-type electrolytes[J].Physical Chemistry Chemical Physics,2014,16(28):14669-14674.
[65] Liu Z,Fu W,Payzant E A,Yu X,Wu Z,Dudney N J,Kiggans J,Hong K,Rondinone A J,Liang C.Anomalous high ionic conductivity of nanoporous β-Li3PS4[J].Journal of the American Chemical Society,2013,135(3):975-978.
[66] Hayashi A,Ohtomo T,Mizuno F,Tadanaga K,Tatsumisago M.Rechargeable lithium batteries,using sulfur-based cathode materials and Li2S-P2S5glass-ceramic electrolytes[J].Electrochimica Acta,2004,50(2-3):893-897.
[67] Hayashi A,Hama S,Morimoto H,Tatsumisago M,Minami T.Preparation of Li2S-P2S5amorphous solid electrolytes by mechanical milling[J].Journal of the American Ceramic Society,2001,84(2):477-79.
[68] Hayashi A,Hama S,Mizuno F,Tadanaga K,Minami T,Tatsumisago M.Characterization of Li2S-P2S5glass-ceramics as a solid electrolyte for lithium secondary batteries[J].Solid State Ion.,2004,175(1-4):683-686.
[69] Sakuda A,Kitaura H,Hayashi A,Tadanaga K,Tatsumisago M.All-solid-state lithium secondary batteries with oxide-coated LiCoO2electrode and Li2S-P2S5electrolyte[J].Journal of Power Sources,2009,189(1):527-530.
[70] Minami K,Hayashi A,Tatsumisago M.Mechanochemical synthesis of Li2S-P2S5glass electrolytes with lithium salts[J].Solid State Ion.,2010,181(33-34):1505-1509.
[71] Minami K,Hayashi A,Tatsumisago M.Characterization of solid electrolytes prepared from Li2S-P2S5glass and ionic liquids[J].Journal of the Electrochemical Society,2010,157(12):A1296-A1301.
[72] Ujiie S,Hayashi A,Tatsumisago M.Structure,ionic conductivity and electrochemical stability of Li2S-P2S5-LiI glass and glass–ceramic electrolytes[J].Solid State Ion.,2012,211:42-45.
[73] Trevey J,Jang J S,Jung Y S,Stoldt C R,Lee S H.Glass-ceramic Li2S-P2S5electrolytes prepared by a single step ball billing process and their application for all-solid-state lithium-ion batteries[J].Electrochemistry Communications,2009,11(9):1830-1833.
[74] Tatsumisago M,Hama S,Hayashi A,Morimoto H,Minami T.New lithium ion conducting glass-ceramics prepared from mechanochemical Li2S-P2S5glasses[J].Solid State Ion.,2002,154–155:635-640.
[75] Minami K,Hayashi A,Tatsumisago M.Crystallization process for superionic Li7P3S11glass-ceramic electrolytes[J].Journal of the American Ceramic Society,2011,94(6):1779-1783.
[76] Muramatsu H,Hayashi A,Ohtomo T,Hama S,Tatsumisago M.Structural change of Li2S-P2S5sulfide solid electrolytes in the atmosphere[J].Solid State Ion.,2011,182(1):116-119.
[77] Ohtomo T,Mizuno F,Hayashi A,Tadanaga K,Tatsumisago M.Electrical and electrochemical properties of Li2S-P2S5-P2O5glass-ceramic electrolytes[J].Journal of Power Sources,2005,146(1-2):715-718.
[78] Minami K,Hayashi A,Ujiie S,Tatsumisago M.Structure and properties of Li2S-P2S5-P2S3glass and glass-ceramic electrolytes[J].Journal of Power Sources,2009,189(1):651-654.
[79] Kennedy J H,Zhang Z M.Preparation and electrochemical properties of the SiS2-P2S5-Li2S glass coformer system[J].Journal of the Electrochemical Society,1989,136(9):2441-2443.
[80] Nagamedianova Z,Sánchez E.Electronic to ionic conductivity of glasses in the Li2S-Sb2S3-P2S5system[J].Solid State Ion.,2006,177(37-38):3259-3265.
[81] PRASADA RAO R ,Seshasayee M.Oxysulfide glasses xLi2O-(1-x)(0.6 Li2S-0.4 P2S5)[J].Journal of Power Sources,2006,159(1):258-262.
[82] Ohtomo T,Hayashi A,Tatsumisago M,Kawamoto K.Suppression of H2S gas generation from the 75Li2S·25P2S5glass electrolyte by additives[J].J.Mater.Sci.,2013,48(11):4137-4142.
[83] Ohtomo T,Hayashi A,Tatsumisago M,Kawamoto K.Characteristics of the Li2O-Li2S-P2S5glasses synthesized by the two-step mechanical milling[J].Journal of Non-Crystalline Solids,2013,364:57-61.
[84] Ong S P,Mo Y,Richards W D,Miara L,Lee H S,Ceder G.Phase stability,electrochemical stability and ionic conductivity of the Li10±1MP2X12(M = Ge, Si, Sn, Al or P, and X = O, S or Se) family of superionic conductors[J].Energy & Environmental Science,2013,6(1):148-156.
[85] Bron P,Johansson S,Zick K,auf der Guenne J S,Dehnen S,Roling B.Li10SnP2S12:An affordable lithium superionic conductor[J].Journal of the American Chemical Society,2013,135(42):15694-15697.
[86] Kuhn A,Duppel V,Lotsch B V.Tetragonal Li10GeP2S12and Li7GePS8-exploring the Li ion dynamics in LGPS Li electrolytes[J].Energy & Environmental Science,2013,6(12):3548-3552.
