MCM-48介孔材料相转化合成的形成机制
2015-09-15袁恩辉邢俊玲逄俊玲姜淑华蒋金刚
袁恩辉 邢俊玲 逄俊玲 姜淑华 蒋金刚*, 张 坤*,
(1华东师范大学化学与分子工程学院,上海市绿色化学与化工过程绿色化重点实验室,上海 200062)
(2南开大学化学学院,先进能源材料化学教育部重点实验室,天津 300071)
M41S系列介孔分子筛材料,包括一维六方孔道结构的MCM-41、三维立方拓扑结构的MCM-48以及层状结构的MCM-50,最早由Mobil公司在1992年研发成功[1-3]。由于该材料具有规整的孔道结构、较大的比表面积(~1 000 m2·g-1)和孔容(1.0 cm3·g-1)以及可以调变的孔径尺寸(2.0~50 nm),在催化、吸附和分离领域引起了人们广泛的研究兴趣。其中,MCM-48具有三维连续的孔道结构,相对于一维孔道的MCM-41介孔材料,在分子扩散方面具有较大的优势和应用前景。但是MCM-48的合成相区狭窄,合成条件比较苛刻,重现性差,从而限制了MCM-48的广泛应用。
文献报道的MCM-48的主要合成策略包括:使用单一的阳离子表面活性剂,如十六烷基三甲基溴化铵 (CTABr),表面活性剂与硅的物质的量之比在0.65~1.5之间,该合成方法成本较高且后处理过程中有大量难于处理的泡沫产生,产物的分离比较困难[4-5];采用混合模板剂的合成策略可以有效地降低阳离子表面活性剂的用量,但是两种模板剂的物质的量之比难于控制,以致于得到的MCM-48介孔分子筛有序性不高[6-7];通过合成新型的表面活性剂,如Gemimi表面活性剂也可以降低表面活性剂的用量,但是该表面活性剂合成程序复杂、产率低,不利于应用于大规模介孔分子筛的合成[8-10];最近张金龙课题组报道了利用F-作为无机矿化剂可以有效合成出高质量的MCM-48介孔材料,但是滤液中大量氟离子的排放对环境是不友好的[11-12]。另外,十六烷基三甲基溴化铵 (CTABr)作为模板剂分子合成的MCM-48介孔材料通常具有水热稳定性差的缺点,550℃焙烧6 h或沸水中回流处理12 h后大部分结构已经塌陷,阻碍了其进一步的相关应用。从上述的报道可见,现有的合成方法存在制备条件苛刻、成本高、污染大以及最终产品的水热稳定性不高等缺点,因此改进MCM-48的现有合成方法是MCM-48制备研究的首要任务。最近我们课题组报道了使用新型的十六烷基三甲基对甲基苯磺酸铵盐(CTATos)作为有机模板剂,可以高效、低成本的合成不同拓扑结构的介孔分子筛,如一维六方结构的MCM-41介孔分子筛、正交结构的SBA-8介孔分子筛、立方结构的MCM-48介孔分子筛以及颗粒尺寸小于100 nm的介孔二氧化硅纳米颗粒(MSNs)[13-17]。该合成策略具有以下优点:表面活性剂的用量低(物质的量之比nCTATos/nSiO2≤0.1),并且产品在抽滤收集过程中不产生大量泡沫,简化了介孔材料的合成工序;通过准确调控反应条件,实现了颗粒大小以及孔径尺寸的精确调控;得到的介孔材料的热和水热稳定性比较高,但是对该类介孔材料合成机理的研究缺乏深入的了解。
在本文中,利用十六烷基三甲基对苯磺酸铵盐(CTATos)作为模板剂成功制备了具有立方结构的MCM-48介孔分子筛,SEM和TEM的表征结果发现:起始相为具有P6mm一维拓扑结构的MCM-41介孔分子筛,随着晶化时间的延长介孔六方相(MCM-41)经立方相 (MCM-48)最终转变成层状相(MCM-50),其中MCM-48为中间相。同时XRD的表征结果证明介孔的孔壁结构随着晶化时间的延长也发生了周期性的变化,由MCM-41和MCM-48原子无序的非晶结构向类沸石分子筛的原子有序结构转化,这个结构转化的合成机制尚未有文献报道。
1 实验部分
1.1 试剂与仪器
试剂:NaOH(AR),天津科密欧化学试剂有限公司;硅溶胶(无钠型,30%),浙江宇达化学试剂公司;十六烷基三甲基对甲基苯磺酸盐 (CTATos),德国MERCK集团公司。
仪器:采用Bruke公司D8 ADVANCE X-射线粉末衍射仪测定样品的晶相结构,Cu靶,Kα辐射源,管电压40 kV,管电流40 mA,λ(Cu Kα1)=0.154 06 nm,λ(Cu Kα2)=0.154 44 nm,比例为 0.497。样品的孔结构采用Quanta chrome公司的Autosorb3-B的自动物理吸附仪进行测定。样品的形貌采用日本日立公司Hitachi S-4800冷场高分辨率发射扫描电子显微镜(SEM)和JEM-2100型透射电子显微镜(TEM)进行测定。TEM测试:所用电子束电压200 kV,样品研细后用乙醇-水制成悬浮液,超声分散后滴加在铜网上进行分析。傅立叶变换红外光谱(FT-IR)采用美国Nicolet公司的Nexus 670型傅立叶变换红外光谱仪测试。29Si魔角旋转固体核磁共振谱在Varian 400-MR核磁共振仪上测得,频率为59.6 MHz,采用直径为4 mm转子,转速为3 kHz,弛豫延迟时间为600 s。
1.2 实验过程
该合成策略与传统介孔分子筛的唯一不同之处就是使用具有较低临界胶束浓度的十六烷基三甲基对甲基苯磺酸铵盐 (CTATos)作为模板剂分子[13-17]。该表面活性剂的阴离子为对甲基苯磺酸根离子(Tos-),而不是常用的Br-离子。Tos-作为阴离子具有如下特点:相对于传统阳离子表面活性剂,CTATos的临界胶束浓度 (critical micelle concentration,简写CMC)较低,从而减少了模板剂的用量;该阴离子具有较大的动力学半径和疏水性,可以有效改变表面活性剂分子的有效堆积常数(即所谓的g值)。典型的合成过程如下:首先将2.10 g NaOH加入到盛有68 mL去离子水的烧杯中,然后加入17.42 g硅溶胶,在60℃下恒温搅拌1 h,得到溶液A;将2.70 g十六烷基三甲基对甲基苯磺酸铵盐(CTATos)加入到盛有80 mL去离子水的烧杯中,60℃下恒温搅拌1 h至溶液变澄清,得到溶液B;将溶液A逐滴加入到溶液B中,继续在60℃下恒温搅拌2 h,得到白色沉淀(该混合物中各物质的物质的量之比为nSiO2∶nCTATos∶nNaOH∶nH2O=1∶0.068∶0.60∶102);将该混合物冷却至室温后,转入聚四氟乙烯反应釜中,在130℃的恒温干燥箱中进行晶化,控制晶化时间,制备得到具有不同拓扑结构的介孔分子筛。所得产物经过抽滤、洗涤、烘干,得到最终产品。
2 结果与讨论
图1为在同一晶化温度,不同晶化时间下所合成的材料的XRD图。图1(a)中,当晶化时间较短时(8 h),存在3个明显的衍射峰,可归属为MCM-41的 3 个特征衍射峰(100),(110)和(200),为一维六方相(P6mm)结构[13]。继续延长晶化时间得到的是MCM-41和MCM-48的物理混合相。当晶化时间延长为24 h,在2θ为2.28°处可以清晰地观察到较强的(211)晶面衍射峰,同时在 2θ=2.15°~3.10°处可以观察到(220)晶面的肩峰,在 2θ=4°~5°处出现多个重叠的衍射峰,说明该样品为具有三维立方结构的MCM-48(Ia3d)介孔分子筛[5-12]。24 h 后,部分 MCM-48相开始转化形成层状相,将晶化时间进一步延长到48 h时,得到的材料可归属为纯的层状的MCM-50介孔分子筛[5]。XRD的结果表明,晶化时间是影响不同介孔相形成的主要因素,并且随着晶化时间的延长,介孔材料由六方相经立方相最终转化为层状相。说明高度有序的MCM-48介孔材料的合成是经过相转变的方式实现的。
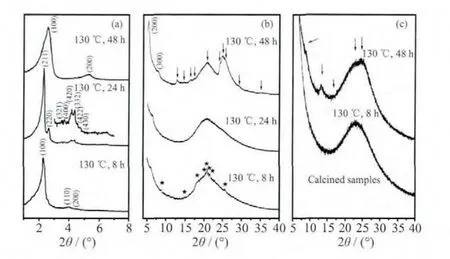
图1 130℃合成温度下改变晶化时间所合成样品的XRD图:(a)原粉的小角XRD,(b)原粉的广角XRD和(c)样品焙烧后的XRD图Fig.1 XRD patterns of samples synthesized at 130 ℃ for varied crystallization time:as-synthesized samples at low-angle range of XRD patterns(a),assynthesized samples at high-angle range of XRD patterns(b),and calcined samples at high-angle range of XRD patterns(c)
通常介孔材料的孔壁具有非晶的结构,即广角XRD不具备特征的衍射峰,只有在2θ为20°时出现一个较宽的无定形二氧化硅特有的“Halo”峰(Halo为“光晕”的意思,在这里特指介孔材料的孔壁具有非晶的无定形结构)。然而,在当前合成样品中,随着晶化时间的延长,样品的广角XRD表明,合成的该系列介孔材料的孔壁结构随着其拓扑结构的转化也发生了明显的改变(图1b)。当晶化时间为8 h,在XRD的高角度区域出现了明显的类似于沸石分子筛的衍射峰,但是强度非常的弱,表明MCM-41孔壁具有一定的原子有序性。随着晶化时间的延长(24 h),即当MCM-48介孔相形成时,在广角XRD上,仅仅在20°左右出现一个单一的“Halo”峰,表明MCM-48的孔壁结构是完全无序的,即介孔材料孔壁的原子有序性完全消失。进一步延长晶化时间(48 h),转化为层状相时,广角XRD图上单一的“Halo”峰完全劈分成2个非常明显的晶体衍射峰,并且2θ在12°~20°之间出现 4个明显的晶体特征衍射峰,表明介孔的孔壁又完全转化为类沸石分子筛的晶体结构,具有非常高的原子有序性。550℃焙烧样品后样品的广角XRD在2θ为12.5°处的衍射峰非常明显(图1c),说明这些层状的类沸石分子筛结构单元具有较高的水热稳定性。总之,XRD的研究结果表明介孔的相转变过程同时伴随着介孔材料孔壁结构由原子无序向有序转变的变化过程。
随着晶化时间的改变,所合成介孔材料的形貌也发生了明显的变化(图2)。晶化时间为8 h时,所得MCM-41介孔材料为管状形貌,且颗粒尺寸主要集中在 2.0~5.0 μm 之间(图 2a)。当晶化时间为 24 h时,MCM-41介孔材料的大部分管状形貌消失,颗粒为不规则的球状,颗粒尺寸集中在200~500 nm之间(图2b),与文献的报道结果完全一致[18]。对该样品SEM图片的仔细观察发现,该样品中仍然存在少量的类似于管状形貌的颗粒 (数理统计的结果显示这些管状形貌的颗粒大约占了总颗粒数的5%),但是基本颗粒尺寸远远小于1.0μm,说明MCM-41向MCM-48相转化的过程中伴随着二氧化硅溶解的过程。但该样品的XRD中不存在任何的MCM-41介孔相的衍射峰,说明这些管状颗粒来不及经历外部形貌转换,直接在颗粒内部由六方结构的MCM-41转化成具有立方结构的MCM-48,由此说明不规则的MCM-48球形颗粒的形成直接来自于管状形貌MCM-41的介孔相,介孔结构的相转变过程可能是通过固-固转化的机理进行的,文献借助于高分辨的透射电镜(HRTEM)表征和结构模拟也证明这个假设[1,19-20]。进一步延长晶化时间至48 h,球形的MCM-48介孔材料完全转化成由纳米片状颗粒聚集而成的橄榄形MCM-50层状相,颗粒高度单分散,且尺寸集中在2.0μm左右。

图2 不同晶化时间下所合成样品的SEM图Fig.2 SEM images of samples synthesized at 130 ℃ for varied crystallization time

图3 不同晶化时间下所合成样品的HR-TEM图Fig.3 High resolution TEM images of samples synthesized at 130℃for varied crystallization time
利用高分辨的透射电镜(HR-TEM)我们对当前介孔材料的孔结构信息也进行了深入的表征(图3)。较短的晶化时间(8 h),得到的介孔材料为典型一维六方结构,介孔的孔径大约为3.0 nm,并且选区电子衍射证明,介孔材料的孔壁为典型的非晶结构(图3a)。而得到的MCM-48介孔材料为典型的三维立方相结构,而且孔结构高度有序,选区的电子衍射证明,孔壁的组成与MCM-41相同,不具有任何的原子有序性(图3b),与广角的XRD表征结果一致。进一步延长晶化时间,MCM-50层状相的孔壁组成发生了明显的变化,选区电子衍射呈现明显衍射斑点,类似沸石分子筛晶体的选区电子衍射花样 (图3c)。我们通过硅烷化扩孔柱撑技术结合红外以及固体核磁共振表征,确定MCM-50的孔壁具有层状硅酸盐Octosilicate的结构,更加详细的实验结果将会后续报道。
通过控制不同的实验条件,不同的课题组发现了介孔材料由六方相(MCM-41)经立方相(MCM-48)向层状相(MCM-50),甚至反向(MCM-50→MCM-48→MCM-41)的转化过程[21-23],但是孔壁由非晶相的原子无序向类分子筛的原子有序的转变过程以及相转化机制还未见文献报道。而此类原子有序的MCM-50介孔分子筛可以作为层状分子筛材料制备扩孔型分子筛的有效前驱体[24]。
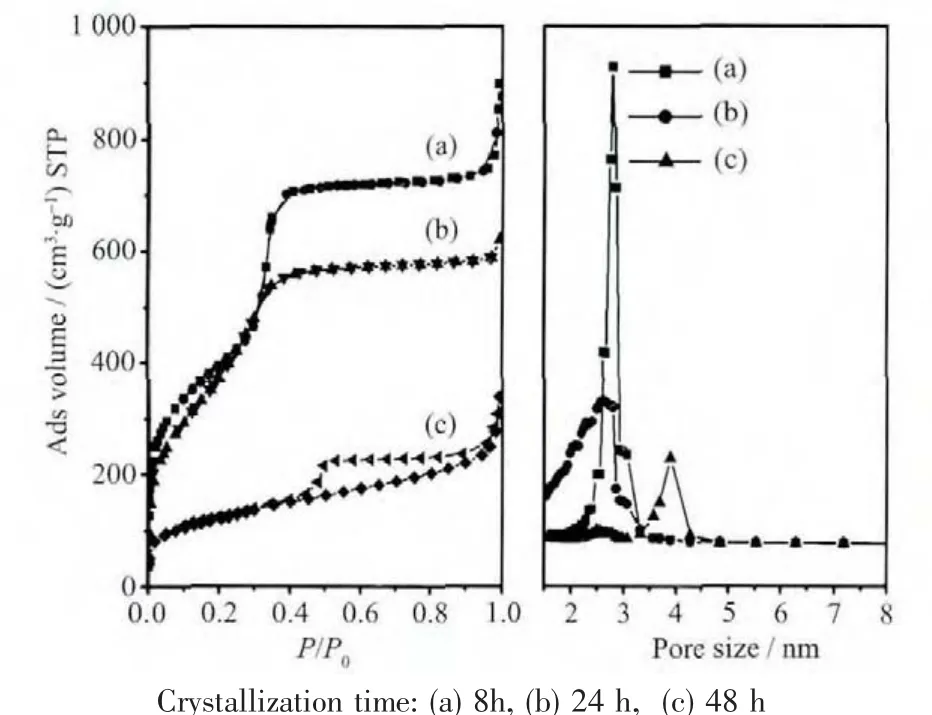
图4 样品焙烧后氮气吸-脱附等温线(左)和BJH孔径分布图(右)Fig.4 N2 adsorption-desorption isotherms(left)and BJH pore size distribution curves(right)of calcined samples synthesized at 130℃for varied
氮气的吸-脱附曲线表明(图4)合成的MCM-48和MCM-41介孔材料为典型的Ⅳ型吸附等温线,具有较高的表面积和孔容(表1)。由于孔道结构高度有序,具有典型的H1型迟滞环结构。MCM-50焙烧以后,由于结构的坍陷,片状结构堆积形成了具有多级孔结构H3迟滞环结构,并且表面积和孔容明显减小,分别为 421 m2·g-1和 0.51 cm3·g-1,与文献报道的结果一致[5]。很明显,仅通过调控晶化时间,利用相转变的合成策略,我们可以比较容易地合成具有不同拓扑结构的介孔分子筛,为介孔分子筛的高效制备提供了一种可以借鉴的方法。
图5所示为不同晶化时间下所合成样品的FTIR谱。在FT-IR谱图高波数区,2 922和2 852 cm-1处的衍射峰为模板剂的特征吸收峰,与文献报道的研究结果一致[13-17]。然而在低波数区400~1 300 cm-1之间,随着晶化时间的延长,红外图谱却发生了明显的变化。当MCM-48介孔相完全转化为MCM-50层状相时,2个新的特征峰在605和1 208 cm-1处的位置出现,并且1 237 cm-1的吸收峰明显变得尖锐,暗示介孔材料的孔壁结构已经发生了明显的变化。低波数区特征吸收峰的出现通常预示着沸石分子筛初级结构单元环状结构的形成[25-27],表明当前MCM-50骨架结构为原子有序的,与前述XRD的表征结果相吻合。另外,更为重要的是在介孔孔道内我们发现了大量表面活性剂阴离子对甲基苯磺酸根离子(Tos-)的存在(825和686 cm-1处),且随着晶化时间的延长Tos-含量明显减小。

表1 不同晶化时间下所合成样品的孔结构信息Table 1 Textural properties of samples synthesized for varied crystallization time

图5 不同晶化时间下所合成样品的FT-IR图谱Fig.5 FT-IR spectrum of samples synthesized at 130℃for varied crystallization time
29Si的固体魔角自旋核磁共振的结果(图6)进一步验证了我们上述的结果,随着晶化时间的延长,硅物种的缩聚程度明显提高,Q3的比例从1.6降到0.9,即表面的硅氧负离子基团(≡SiO-)明显减少。并且在MCM-50形成以后,Q3和Q4完全劈分成2个尖锐的吸收峰,说明MCM-50结构中硅物种的环境发生了明显的变化。实际上,只有传统沸石分子筛材料才具有类似的吸收峰,即当前合成的MCM-50的孔壁结构呈现一定的原子有序性。因此结合红外和固体Si核磁的表征结果以及表面活性剂的有效堆积参数g(g=V/(a0l)),V代表表面活性剂分子的链及链间助溶剂所占的总体积,a0代表表面活性剂极性头所占的有效面积,l代表表面活性剂长链的有效长度;当g在1/3至1/2之间生成MCM-41,1/2到2/3之间生成MCM-48,接近1时生成MCM-50层状相。),我们推断MCM-48的形成机制如下:由于体积较大的对甲基苯磺酸根离子(Tos-)具有较强的疏水性,对阳离子表面活性剂分子具有较强的结合作用,在表面活性剂胶束和无机硅界面形成了阴阳离子对{CTA+Tos-},反应开始时,大量体积较大的Tos-物种存在,由于空间效应表面活性剂极性头所占的有效面积a0明显增加,即g值减小,容易形成具有P6mm的二维六方相;随着反应时间的延长,Tos-物种容易流失在孔道中的量逐渐减小,从而a0随之减小,导致g值变大,最终经Ia3d立方相向层状相结构转化。

图6 不同晶化时间下所合成样品的固体29Si MAS NMR图谱Fig.6 Solid 29Si MASNMR spectrum of samples synthesized at 130℃for varied crystallization time
3 结 论
以十六烷基三甲基对甲基苯磺酸铵盐(CTATos)为模板剂,通过水热合成的策略实现了具有不同拓扑结构纯硅介孔分子筛的合成。X-射线衍射、高分辨扫描电子显微镜、透射电子显微镜、N2吸-脱附实验、Fourier变换红外光谱和29Si的固体魔角自旋核磁共振等测试手段对所合成材料的研究结果表明:三维立方拓扑结构的MCM-48介孔分子筛形成是通过相转变的合成方式进行的,并且最终得到MCM-50层状相,具有类沸石分子筛的原子有序结构。结合表面活性剂的有效堆积参数g推断了该相转变发生的形成机制。我们最新的研究结果表明原子有序的MCM-50是一种新型的层状结构沸石分子筛前驱体,利用后合成改性的方法,如层间柱撑,可以合成扩孔型的新型沸石分子筛,相关研究工作正在开展当中。
[1]Wan Y,Zhao D Y.Chem.Rev.,2007,107(1):2821-2860
[2]Wan Y,Yang H F,Zhao D Y.Accounts.Chem.Res.,2006,39(7):423-432
[3]Hsueh H Y,Yao C T,Ho R M.Chem.Soc.Rev.,2015,44(7):1974-2018
[4]Vartuli J C,Schmitt K D,Kresge C T,et al.Chem.Mater.,1994,6(12):2317-2326
[5]Beck JS,Vartuli JC,Roth W J,et al.J.Am.Chem.Soc.,1992,114(27):10834-10843
[6]LIU Chun-Yan(刘春艳),RONG Zhi-Hong(荣志红),WANG Xiao-Qing(王小青).Chinese J.Inog.Chem.(无机化学学报),2008,24(7):1068-1072
[7]Chen FX,Song FB,Li QZ.Microporous Mesoporous Mater.,1999,29(3):305-310
[8]HU Jun(胡军),ZHOU Li-Hui(周丽绘),LI Hong-Ning(李鸿宁),et al.Acta Phys.-Chim.Sin.(物理化学学报),2005,21(11):1217-1222
[9]Han S H,Xu J,Hou W G,et al.J.Phys.Chem.B,2004,108(39):15043-15048
[10]Huo Q S,Margolese D I,Stucky GD.Chem.Mater.,1996,8(5):1147-1160
[11]Wang L Z,Zhang JL,Chen F,et al.J.Phys.Chem.C,2007,111(37):13648-13651
[12]Shao YF,Wang LZ,Zhang JL,etal.Microporous Mesoporous Mater.,2005,86(1/2/3):314-322
[13]Zhang K,Chen H L,Albela B,et al.Eur.J.Inorg.Chem.,2011,1:59-67
[14]Zhang K,Zhang Y,Hou QW,et al.Microporous Mesoporous Mater.,2011,143(2/3):401-405
[15]Zhang K,Yuan E H,Xu L L,et al.Eur.J.Inorg.Chem.,2012,26:4183-4189
[16]Zhang K,Xu L L,Jiang JG,et al.J.Am.Chem.Soc.,2013,135(7):2435-2430
[17]Yu YJ,Xing JL,Pang JL,et al.ACSAppl.Mater.Interfaces,2014,6(24):22655-22665
[18]Sun H J,Coppens M O.J.Mater.Chem.,2002,12:3016-3020
[19]Omer L,Ruthstein S,Goldfarb D,et al.J.Am.Chem.Soc.,2009,131(34):12466-12473
[20]Han L,Miyasaka K,Terasaki O,et al.J.Am.Chem.Soc.,2011,133(30):11524-11533
[21]Díaz I,Pérez-Parientea J,Terasaki O.J.Mater.Chem.,2004,14:48-53
[22]Che S A,Kamiya S,Terasaki,O,et al.J.Am.Chem.Soc.,2001,123(48):12089-12090
[23]Matijasic A,Voegtlin A C,Patarin J,et al.Chem.Commun.,1996:1123-1124
[24]Wu P,Ruan JF,Wang L L,et al.J.Am.Chem.Soc.,2008,130(26):8178-8187
[25]Christiansen S C,Zhao D Y,Janicke M T,et al.J.Am.Chem.Soc.,2001,123(19):4519-4529
[26]Hedin N,Graf R,Christiansen S C,et al.J.Am.Chem.Soc.,2004,126(30):9425-9432
[27]Zhang K,Wang Y M,Albela B,et al.New J.Chem.,2009,33:2479-2485