耐硫甲烷化反应的研究进展
2015-08-20王玮涵李振花王保伟徐艳马新宾
王玮涵,李振花,王保伟,徐艳,马新宾
(天津大学化工学院,绿色合成与转化教育部重点实验室,天津 300072)
引 言
能源和环境是当今社会最为关注的焦点之一。石油和天然气作为全球能源消耗和化工原料的主要来源,其产量却难以匹配日益增长的能源需求。中国的能源结构特点是“富煤、贫油、少气”,煤炭在我国的能源中占据着主导位置,因此利用相对丰富的煤炭资源发展煤制天然气产业,是缓解我国天然气供求矛盾的一条有效途径。天然气主要成分是甲烷。与相同质量的煤炭相比,天然气燃烧排放的CO2仅为煤炭的40%,没有废水、废渣和粉尘产生。作为热值高、效率高、污染小的优质能源,天然气具有其他燃料不可比拟的优势。
甲烷化技术是煤制天然气工艺的核心技术之一。传统过程通常采用间接甲烷化工艺,如图1 所示[1],以Ni[2-5]、Co[6-7]或贵金属Ru[8-9]等为活性组分,其中Ni 基催化剂已在工业化中广泛应用[10]。此工艺路线的优点在于处理量大、技术路线较为成熟,国内外已商业化和正在中试的工艺过程均采用间接甲烷化工艺路线[1]。但是,该体系的催化剂对硫组分极为敏感,原料气需要先精脱硫至浓度低于0.1×10-6;为了满足工艺要求,原料气还需经水汽变换反应将H2/CO 比例调整为3 以上。这两点在很大程度上限制了Ni 基催化剂的应用。因此,开发在低H2/CO比例的含硫气氛中仍具有较好甲烷化活性的催化剂,是甲烷化技术的发展方向。
美国煤气研究所最早开发了耐硫甲烷化工艺,即直接甲烷化工艺[11],该工艺集耐硫甲烷化和水汽变换于一体,如图2 所示。在耐硫甲烷化工艺中,原料气无须脱硫,且无须加入水蒸气提高H2/CO比,因此工艺流程得到极大简化,进而节省了投资并降低了能耗。目前,耐硫甲烷化催化剂主要为Mo 基催化剂,在含硫气氛中的催化反应活性相为MoS2[12]。MoS2在加氢脱硫[13-16]、加氢脱氮[17]、加氢脱氧[18]等含硫气氛的加氢反应体系中得到广泛的应用。此外,此类催化剂兼备良好的水汽变换性能[18-19]。因此,Mo 基催化剂在低H2/CO 比例的耐硫甲烷化反应中具有明显的优势。
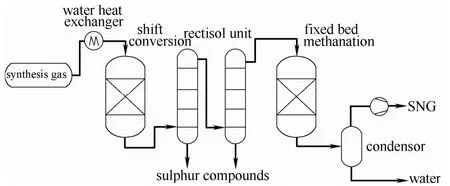
图1 间接甲烷化工艺简单流程[1]Fig.1 Simple flowchart of indirect methanation process[1]
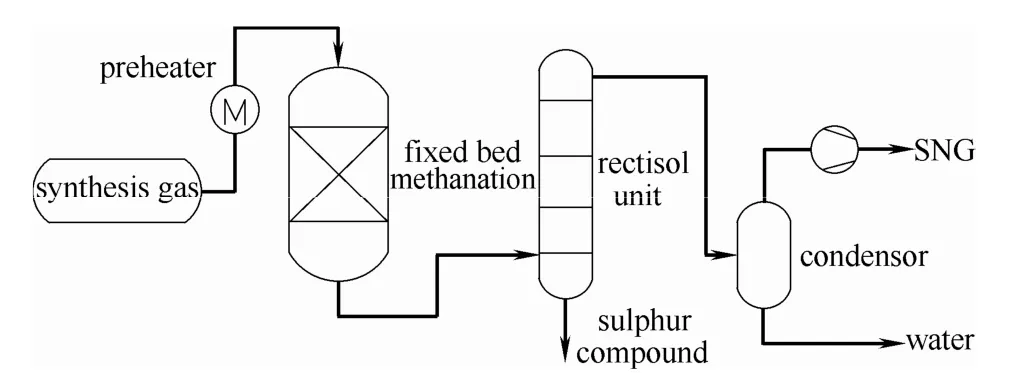
图2 直接甲烷化工艺简单流程Fig.2 Simple flowchart of direct methanation process
间接甲烷化反应工艺中的CO 甲烷化反应是在H2/CO 比例不低于3 的条件下按以下反应式进行的。
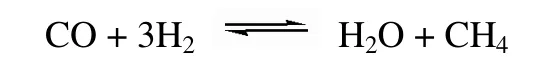
为满足工艺要求,原料气需先经水汽变换反应来提高H2/CO 比例。
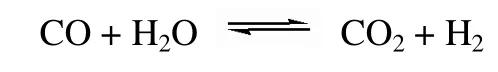
而在直接甲烷化中,甲烷化和水汽变换两个反应同时在Mo 基催化剂上发生,因此甲烷化反应可直接描述为
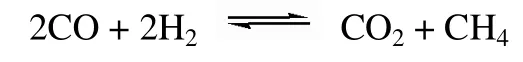
从该反应式中也可看出,直接甲烷化工艺符合低H2/CO 比例的原料气组分要求。CO 甲烷化反应和水汽变换反应在热力学上是可行的[20],该工艺技术的关键问题在于开发在较宽温度范围(300~650℃)兼具高活性和稳定性的耐硫甲烷化催化剂。
关于间接甲烷化催化剂的研究进展已有诸多报道[1,10,21-23]。胡大成等[22]和Gao 等[10]分别介绍了间接甲烷化催化剂及其失活机理、间接甲烷化的反应机理等研究进展;张成[23]着重介绍了Ni 基间接甲烷化催化剂的研究进展;Wang 等[24-25]对CO2甲烷化催化剂、反应机理和反应器的研究进行了综述;崔凯凯等[26]则针对CO2甲烷化催化剂的载体和助剂对催化性能的影响进行了介绍。以往的综述均侧重于间接甲烷化工艺及其催化剂体系,鲜见针对直接甲烷化体系的耐硫催化剂的评述。本文全面地介绍了耐硫甲烷化催化剂制备及其硫化机理、反应机理等方面的研究进展,这将对今后耐硫甲烷化体系的研究提供帮助。
1 Mo 基耐硫甲烷化反应催化剂
耐硫甲烷化催化剂主要是以MoS2为活性组分的负载型催化剂。MoS2一般由氧化物在一定比例的H2S/H2气氛中进行还原硫化得到。催化性能受到前驱体、载体和助剂种类的影响。
1.1 活性组分前驱体对催化剂性能的影响
常见的Mo 基催化剂的前驱体有仲钼酸铵、四硫代钼酸铵等。仲钼酸铵作为前驱体经焙烧可到氧化钼,再经硫化可形成堆叠层数较多的MoS2层状结构。四硫代钼酸铵在惰性气氛中可直接分解形成非化学计量的MoS2,不仅可以省去硫化过程,还能减弱Mo 与载体之间的强相互作用[27]。但是,仲钼酸铵作为前驱体经焙烧和硫化处理得到的MoS2以无定形为主,而四硫代钼酸铵在惰性气氛中分解主要形成晶态MoS2,无定形MoS2甲烷化活性高于晶态MoS2[28]。再者,四硫代钼酸铵价格昂贵。与之相比,仲钼酸铵在实验研究和工业生产中均有更好的应用前景。
1.2 载体对催化剂性能的影响
载体能够有效地负载和分散活性组分,提高催化剂的效率,降低催化剂的成本,并且常与活性组分共同对催化反应产生作用,影响催化剂的活性、稳定性和产物选择性。Kim 等[29]在500℃条件下考察了不同载体负载的Mo 基催化剂的耐硫甲烷化活性,按以下顺序递减:YSZ > γ-Al2O3> ZrO2> CeO2> TiO2> SiO2> SiO2-Al2O3。伏义路等[30]研究了不同载体上MoS2催化剂的甲烷化反应性能,得到400℃ 时甲烷化活性顺序为:ZrO2> γ-Al2O3> CeO2> La2O3,热稳定性顺序为:La2O3> CeO2> γ-Al2O3> ZrO2。秦绍东等[31]对比了γ-Al2O3、CeO2和ZrO2负载的Mo 基催化剂性质及其甲烷化性能,结果表明ZrO2载体上MoO3分散度最高,而CeO2载体的相抗烧结能力最强。Wang 等[32]则对CeO2-Al2O3、MgO-Al2O3、TiO2-Al2O3、ZrO2-Al2O3复合载体负载的 CoO-MoO3催化剂进行了活性比较,其中CeO2-Al2O3复合载体表现出较好的催化活性,这归因于CeO2的添加能够促进活性组分MoS2的分散。
1.2.1 γ-Al2O3载体 γ-Al2O3载体具有较大的比表面积、丰富的孔隙率和良好的机械强度,在工业上有广泛的应用。MoO3在γ-Al2O3载体上的分散阈值约为4.04 atom/nm2[33-34]。Wang 等[35]认为当MoO3负载量低于分散阈值时,Mo6+以无定形形式存在,为四面体配位和八面体配位物种,如图3 所示,其中四面体配位Mo6+物种易于形成不饱和配位的Mo离子,是甲烷化活性位的前驱体;当MoO3负载量高于分散阈值时,过量的MoO3在低于700℃ 时会团聚形成晶态,从而降低甲烷化活性。随着负载量的增加,MoO3晶粒尺寸增大,并与Al2O3形成Al2(MoO4)3物种,Al2(MoO4)3物种的生成消耗活性Mo 组分,且Al2(MoO4)3中的Mo 难以被硫化,不利于甲烷化反应。文献中对Al2(MoO4)3物种的形成条件见解不一:Li 等[36]认为当MoO3负载量达到一定值即可形成Al2(MoO4)3物种;但是,也有文献认为Al2(MoO4)3物种形成需同时具备两个条件:高温(>700℃)和MoO3负载量超过单层分散阈值[37],但其温度要求与Wang 等[35]的实验结果并不完全一致。
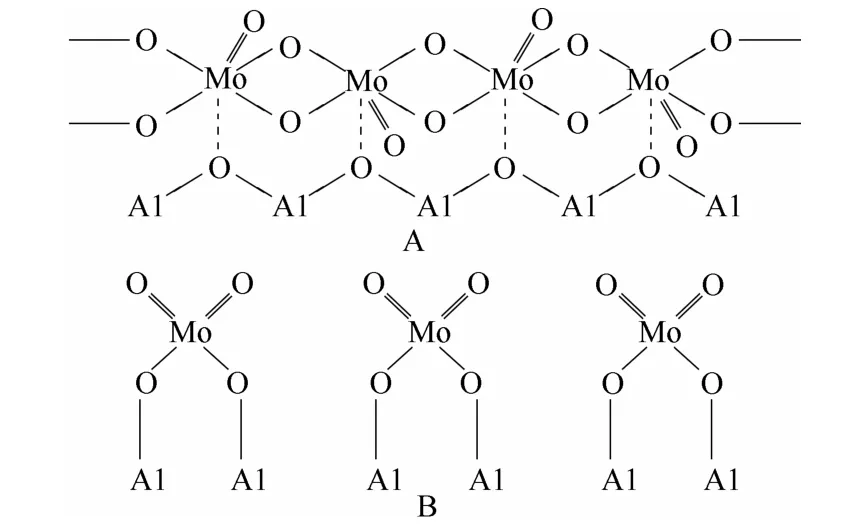
图3 八面体配位(A)和四面体配位(B)的Mo6+物种Fig.3 Octahedrally coordinated Mo6+ (A) and tetrahedrally coordinated Mo6+ (B)
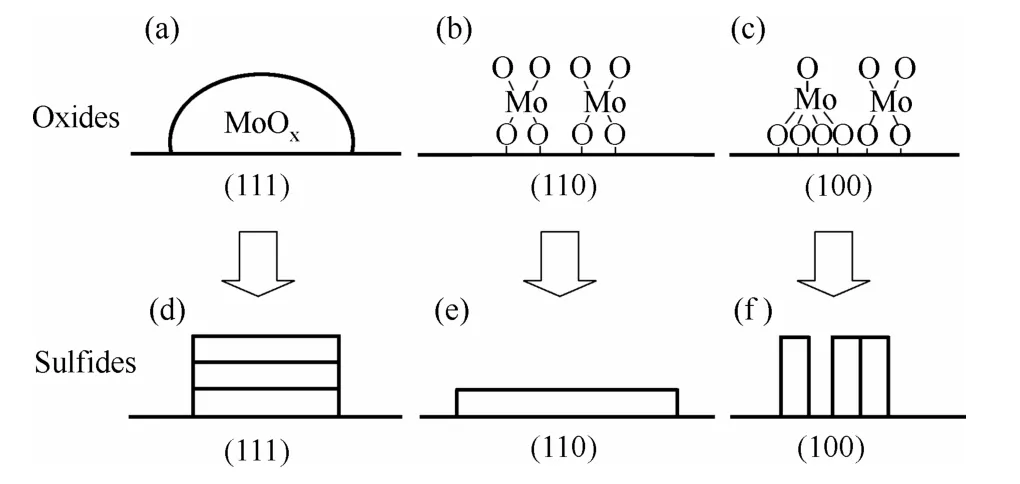
图4 经773 K 硫化后Al2O3 不同晶面上Mo 的氧化物和 硫化物的微观结构[38]Fig.4 Schematic diagram of morphologies of MoOx and MoS2 clusters after sulfidation at 773 K on different planes of Al2O3[38]
γ-Al2O3表面的物理化学性质对活性组分的分散和负载有显著影响。Sakashita 等[38-39]发现γ-Al2O3载体的表面取向影响了Mo 的氧化态和硫化态的微观结构和分散度。片状γ-Al2O3的表面取向主要是{110}晶面,负载的MoS2多为具有单层条纹结构的无定形MoS2;球形γ-Al2O3的表面取向主要是{100}和{111}晶面,MoS2多以高度分散或具有多层堆垛结构的无定形态分布,如图4 所示。姚玉芹等[40]认为当活性组分负载量低于分散阈值时,Al2O3载体的比表面积与催化剂的活性无直接关系,但是集中的孔分布有利于催化剂活性的提高;随着载体单位面积酸量的增加,甲烷化活性呈现先增大后减小的规律。Chen 等[41]对Al2O3表面的酸位和酸量进行调变,发现Al2O3表面酸性主要影响了MoS2的电子特性,对MoS2的形态和分散影响不大。目前,针对Al2O3载体表面性质与耐硫甲烷化活性的关系的报道还较少。
1.2.2 ZrO2载体 ZrO2载体是兼具酸碱性和氧化还原性的过渡金属氧化物,已在间接甲烷化、水汽变换、加氢脱硫、加氢脱氮等催化研究中引起了广泛的关注。Li 等[42]制备了 25%(质量分数)MoO3/ZrO2催化剂用于耐硫直接甲烷化反应,在550℃ 时CO 转化率可达到90.06%,接近于该条件下的CO平衡转化率并明显高于MoO3/Al2O3的催化活性,这是由于MoO3在ZrO2载体上分散度高且容易被还原。
ZrO2载体能够和活性组分产生强烈的相互作用。当MoO3的负载量低于其在ZrO2载体上的单层分散阈值5.0 atom/nm2时,单斜晶型ZrO2(m-ZrO2)比四方晶型(t-ZrO2)能够更有效地分散MoO3,避免了晶相MoO3的出现;随着负载量的增加,MoO3促使载体从m-ZrO2向t-ZrO2转变[42],这种相转变归因于MoO3负载导致的晶体边界区域的减小[43];当负载量超过单层分散阈值或焙烧温度高于600℃ 时,载体表面的MoO3晶态化,并与四方晶型形成Mo(ZrO4)2物种[44],从而导致甲烷化活性下降[42];当焙烧温度继续升高至800℃时,Mo(ZrO4)2分解生成晶态的MoO3和m-ZrO2[43]。ZrO2载体晶型往往对负载的活性中心的结构、催化活性和选择性有显著影响。Yamasaki 等[45]发现ZrO2负载的Ni 基催化剂的CO2甲烷化活性随t-ZrO2含量的增加而提高;Rhodes 等[46]的实验结果表明在CO 加氢制甲醇的反应中,Cu/m-ZrO2催化剂上的甲醇生成速率约为Cu/t-ZrO2的34 倍;He 等[47]发现在CO 加氢制备醇类的反应中ZrO2的晶型对碳链增长概率有一定影响,Pd/m-ZrO2上的主产物为异丁醇,在Pd/t-ZrO2上的主产物为乙醇。目前尚无ZrO2的晶型效应对耐硫甲烷化催化剂性能影响的相关报道,开展相关的研究有助于认识MoO3/ZrO2催化剂中载体和金属氧化物的相关作用及其对催化剂构效关系的影响。
1.2.3 CeO2-Al2O3载体 稀土金属氧化物CeO2具有n 型半导体性质,常作为催化剂的载体或助剂。随着氧化还原气氛的不同,Ce 在催化剂中以Ce4+/Ce3+价态存在,在结构中易形成流动的空位,因此具有较好的氧储存性能和氧表面传递能力,并能与金属界面产生协同作用,有利于提高催化剂的活性[48]。Tada 等[49]发现Ni/CeO2催化剂用于间接甲烷化体系,CeO2表面的还原性显著促进了CO2甲烷化反应;CeO2作为载体不仅可以提高甲烷化反应活性,还明显改善了催化剂的抗积炭能力[50-51]。但是,CeO2作为载体时比表面积很小,而采用特殊方法获得的大比表面积的纳米CeO2颗粒载体机械强度和耐高温性能差,在高温下易发生结构塌陷并烧结[52]。因此获得兼具高比表面积、良好力学性能和耐高温性能的CeO2载体是研究的重点。
将CeO2作为载体助剂掺杂在Al2O3中形成CeO2-Al2O3复合载体,能够明显改善活性组分的分散并提高催化剂的稳定性,同时也避免了CeO2单独作为载体时耐高温性能差的缺点。Wang 等[53]考察了CeO2-Al2O3复合载体制备方法对其性质和催化活性的影响,认为具有较大的比表面积、较小的CeO2晶粒和较弱的Ce-Al 相互作用,有利于甲烷化活性的提高。文献中采用沉积沉淀法制备CeO2-Al2O3复合载体,发现Al2O3中基质表面形成一层CeO2,负载MoO3后部分MoO3直接与CeO2接触[54-55],如图5 所示。由于MoO3与CeO2之间的相互作用弱于MoO3与Al2O3之间的相互作用,MoO3负载在CeO2-Al2O3复合载体上更容易被还原。Jiang 等[56]采用共沉淀法制备MoO3/CeO2-Al2O3催化剂,发现部分Ce3+向Al2O3晶格结构中迁移形成针状的CeAlO3物种,从而提高催化剂的稳定性。

图5 MoO3/CeO2-Al2O3 催化剂模型[55]Fig.5 Proposed model of MoO3/CeO2-Al2O3 catalyst[55]
1.3 助剂对催化剂性能的影响
助剂的加入可以改善活性组分在载体表面的分散度、氧化还原能力以及活性位的结构,从而改善催化剂的活性和稳定性等。Sasaki 等[57]在水汽变换反应中考察了助剂对硫化态的MoO3/Al2O3催化剂的影响,助剂对催化活性的影响按以下顺序排列:Ni>Co>Ti>Zr>Zn>Mg>Ca,这一顺序与元素的电负性大小一致,即电负性较大的助剂可促进MoO3的还原和硫化度,从而提高催化剂的活性。Chen 等[58]通过DFT 计算关联了助剂的电负性和价电子数与其作为助剂对MoO3的还原和硫化的影响,并得到相同的规律。
Co 和Ni 作为结构助剂,能够修饰MoS2的层间和边缘位点,取代边缘的Mo 原子,很大程度上减小催化剂边缘S 的键能并降低边缘S 的覆盖度,促使催化剂边缘表面产生了更多的配位不饱和空位[59]。助剂的含量影响CoMoS 或NiMoS 活性位的数量,进而影响到催化剂的加氢处理能力[60-61]。Co和Ni 助剂的加入虽然对Mo 基耐硫甲烷化催化剂的活性具有很好的促进效果,但是也增强了碳链增长能力,降低了CH4选择性[62]。另外,Ni 助剂的加入也伴随有与Ni 基催化剂相同的积炭问题,随着活性位被积炭所覆盖,催化剂稳定性变差[63]。
2 Mo 基催化剂的硫化过程及机理
2.1 Mo 基催化剂的硫化方法
Mo 基耐硫甲烷化催化剂的硫化方法有两类:一类是在线硫化,即硫化与反应为同一反应器,硫化完毕的催化剂直接用于催化反应;另一类是离线硫化,即硫化与反应过程分离,硫化完毕的催化剂重新装填进反应器进行反应。离线硫化的催化剂在装填时会接触空气,于催化剂的活性不利;而在线硫化可以避免这一缺点,且操作相对简单。
常见的硫化剂有固相的硫粉、液相的CS2或多硫化物溶液、气相的H2S 或含H2S 的原料气等。相比而言,固态和液态硫化剂的硫化能力较强,气态硫化剂相对较弱。固态硫化剂多用于离线硫化;液态硫化剂在两种方式中均有应用,由于其来源广泛、价格低廉,在工业硫化中已应用普遍,但是部分硫化剂,如CS2等,因毒性较大或刺激性较强存在一定的限制;气体硫化剂因其使用方便,并易于处理,在工业上的在线硫化方式中有广泛应用。在耐硫甲烷化反应体系中,通常采用含一定量H2S 的H2气氛对Mo 的氧化物进行硫化和还原[54-56,60-61],此外也有关于硫粉预硫化Mo 基催化剂的相关报道[64]。
2.2 Mo 基催化剂硫化机理
Mo 基催化剂在含硫气氛的甲烷化反应的活性相为MoS2,由MoO3硫化生成MoS2是一个复杂的化学反应过程,其反应机理目前尚未达成共识,分歧主要在于H2开始参与反应的阶段及其作用的对象,可能的反应途径由图6 所示。
Arnoldy 等[65]认为H2在MoO3的硫化过程中起主要作用,且空间体积较大的S 原子对MoO 键上S 取代O 的过程产生位阻效应,导致MoO3中的3 个O 全被S 取代的可能性很小,因此硫化过程机理应按图6 所示的途径Ⅰ→Ⅱ→ Ⅴ → Ⅵ→Ⅶ 或 Ⅰ →Ⅴ →Ⅵ →Ⅶ 进行。同时指出,MoO2和Mo的硫化需要在高温下进行,尽管前处理条件会有所影响,但MoO3完全硫化的温度在800℃以上,这与Farag[66]得到的结论基本一致。
De Boer 等[67]则认为H2在硫化过程中的作用较小,通过考察硫化产物结构和S/Mo 摩尔比的影响,发现MoO3经S-O 交换形成氧硫钼化合物MoOxSy,随温度升高进行深度硫化生成类似MoS3结构的物质,进一步被H2还原为MoS2,即硫化过程机理为图6 所示的Ⅰ → Ⅱ →Ⅲ → Ⅳ →Ⅶ 途径。De Boer 的硫化机理是针对MoO3/SiO2催化剂的硫化研究的基础上提出的,它与MoO3/Al2O3催化剂的硫化机理基本相同,且SiO2作为载体可减小载体与Mo 之间的相互作用,更有利于硫化机理的研究[68]。
Weber 等[69]采用XPS 和IRES 技术对MoO3的硫化和(NH4)2MoO2S2的分解过程进行研究,认为硫化机理更可能按图6 所示的途径Ⅰ→Ⅱ →Ⅲ → Ⅶ 进行。但是,这一理论认为MoOS2中间态化合物中MoO 上的O 位于端位,而Shi 等[70]已证明MoO3上端位的O 最容易被S 取代,在硫化过程中保持端位MoO 的存在非常困难,因此,该硫化过程机理仍值得商榷。
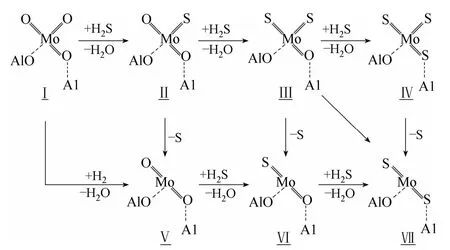
图6 MoO3 的硫化机理Fig.6 Sulfidation mechanism of MoO3
2.3 Co 和Ni 助剂对硫化过程的影响
前文已提到,在Mo 基耐硫甲烷化催化剂中加入Co 和Ni 助剂,可以生成Co(Ni)-Mo-S 活性结构。Ratnasamy 等[71]最早提出Co 与MoS2形成类似四面体金字塔的CoMoS 结构,之后Topsøe 等[72-73]又对这一结构模型进行详细的阐述,现在CoMoS 结构已得到普遍认同。对于Co 在MoS2上的存在形式,Lauritsen等[74]认为Co原子取代了MoS2结构边缘位的Mo,但也有学者认为Co 仅是在MoS2的边缘位S 上起修饰作用[73,75]。CoMoS 结构使得催化剂表面出现溢流效应,为催化反应过程提供了所需要的氢离子,从而提高了催化剂的加氢反应性能[75]。
CoMoS 活性结构形成的关键因素为催化剂中Co/Mo 比例和硫化温度[76-77]。其中,催化剂的最佳Co/Mo 比与载体种类和反应体系有关[76,78-79],而硫化温度是影响CoMoS 活性结构形成的主要因素。文献报道CoMoS 活性结构有两种形式:CoMoS(Ⅰ)型和CoMoS(Ⅱ)型,CoMoS(Ⅰ)型通常在400℃ 左右形成,而形成CoMoS(Ⅱ)型的硫化温度约600℃。在耐硫甲烷化反应体系中,CoMoS(Ⅰ)型的活性明显高于CoMoS(Ⅱ)型,CoMoS(Ⅱ)活性较差的原因是在形成过程中伴随着大量的Co9S8和MoS2晶体形成,不利于耐硫甲烷化反应[60,80]。在添加Co 助剂的Mo 基催化剂中,活性组分的存在形式主要有高度分散的CoO 和MoO3,以及α-CoMoO4和β-CoMoO4物种[81]。不同温度下CoO-MoO/γ-Al2O3催化剂的硫化行为如图7 所示[60]:β-CoMoO4经硫化后生成CoMoS 活性结构,而α-CoMoO4的硫化产物以Co9S8和MoS2为主;CoMoO4在300℃即可硫化完全,在400℃硫化温度下主要得到CoMoS(Ⅰ);随着硫化温度的继续升高,CoMoS 结构逐渐由CoMoS(Ⅰ)型向CoMoS(Ⅱ)型转变;当硫化温度高于500℃,Co9S8和MoS2晶体逐渐形成。文献中报道的NiMoS 活性结构的耐硫甲烷化性能及硫化温度对其影响与CoMoS 结构一致[61]。

图7 不同温度下CoO-MoO/γ-Al2O3 催化剂硫化行为[60]Fig.7 Schematic representation of sulfidation process of CoO-MoO/γ-Al2O3 catalysts at different temperatures[60]
2.4 CeO2 的硫化行为
在CeO2-Al2O3复合载体负载的MoO3催化剂中,CeO2对Al2O3表面起修饰作用,改善了催化剂上Mo 物种与载体之间的相互作用。同时,CeO2在硫化气氛中还表现出一些特殊的性质。Kempegowda 等[82]发现CeO2在500~800℃ 之间可以被硫化生成Ce2O2S 物种,这种物种在更高温度及含氧气氛中能够再次氧化为CeO2。
Jiang 等[55]研究了硫化温度对 MoO3/CeO2- Al2O3催化剂的影响,在硫化条件下CeO2主要有两个反应路径:(1)Ce4+被H2还原为Ce3+;(2)与H2S 生成Ce 的含硫化合物。当温度高于500℃时,CeO2能够较好地还原为Ce2O3物种,进而被硫化生成Ce2O2S。同时,部分Ce3+向Al2O3晶格结构中迁移(和/或Al3+向Ce2O3晶格结构中迁移)从而形成CeAlO3物种。换言之,MoO3/CeO2-Al2O3催化剂表面形成的CeO2层随着硫化温度的升高逐渐消失。Ce 的硫化和迁移破坏了催化剂表面的CeO2,但是增强了它与Al2O3之间的相互作用,因此催化剂活性下降的同时稳定性得以改善。Jiang 等[83]对比了分步硫化对MoO3/Al2O3催化剂和MoO3/CeO2-Al2O3催化剂的影响,认为分步硫化的作用对象是Ce 物种而非Mo 物种。分步硫化方式降低了高温硫化对CeO2的破坏,使更多的CeO2保留于催化剂表面,从而提高了催化剂的活性。
3 耐硫甲烷化反应机理
CO 甲烷化的反应机理已有大量的研究报道,但是目前尚未达到共识。根本分歧在于CO 是直接解离还是氢助解离,以及CO 甲烷化的速控步骤是CO 的解离还是表面碳的加氢。相关的CO 甲烷化反应机理方面的报道大多是基于较为成熟的Ni 基催化剂或贵金属催化剂体系[10,22]。关于耐硫甲烷化体系,范崇正等[84]在脉冲反应装置对MoS2催化甲烷化反应进行考察,认为该反应很可能是通过表面活化配合物进行的,即CO 与H2吸附在催化剂样品上先形成M-CHOH 的烯醇式结构,然后继续与H2反应生成甲烷化,这可能是Mo 基催化剂与Ni 基催化剂对于CO 甲烷化反应催化机理的重大差别。Shi等[85]采用双S-Mo-S 薄层模型对Al2O3负载MoS2催化剂(10-10)晶面上的CO 加氢反应进行DFT 计算,认为CO 吸附在MoS2表面边缘的桥联Mo 上并形成倾斜的结构,不可能直接解离为C 和O;MoS2簇上吸附的一些C1 型表面物种可能是CO 加氢反应的中间产物,CO 加氢合成CH4的过程按照以下机理进行:CO → CHO → CH2O → CH2OH → CH2→ CH3→ CH4。目前对于耐硫甲烷化反应机理还缺乏深入的研究探讨,仍需针对反应过程的细节做进一步阐明。
4 结语和展望
综上所述,虽然目前已有很多关于Mo 基催化剂加氢反应体系的研究报道,包括催化剂的制备和硫化等,相关结论均可在耐硫直接甲烷化体系的研究中进行借鉴,但是针对Mo 基耐硫直接甲烷化催化剂的研究相对较少,尤其是关于较低反应温度下仍具有较高催化活性的催化剂研发和耐硫直接甲烷化反应机理方面。今后,耐硫直接甲烷化体系的研究工作,不仅需要从理论角度探讨反应机理和催化剂开发,而且需要针对甲烷化反应强放热的特点对工艺技术进一步研究。研究的重点包括以下方面。
(1)低温条件下耐硫甲烷化催化剂活性的提高。已见报道的耐硫直接甲烷化催化剂大多是在500~600℃ 范围内具有较好的催化活性,这一温度范围明显高于间接甲烷化的反应温度范围。因此,提高催化剂在低温条件下的催化剂活性,增大其活性温度范围,具有重要的科学和现实意义。
(2)高温条件下甲烷化反应和水汽变换反应的耦合。水汽变换反应在高温条件下的反应速率很快。在较高的反应温度范围内,不仅要针对催化剂的热稳定性能和抗烧结性能进行研究,而且需要进一步提高耐硫直接甲烷化催化剂的活性,并使之与水汽变换反应速率相匹配。
(3)耐硫直接甲烷化反应机理的研究。目前,CO 甲烷化反应和水汽变换反应机理虽然仍存在分歧,但已有一些重要的成果。然而,针对不同催化体系的作用机理缺乏深入的探讨。在耐硫直接甲烷化反应体系,需要针对Mo 基催化剂进一步研究催化剂的作用机理、中间产物物种和反应途径、催化剂活性降低甚至失活的反应机理。
(4)耐硫直接甲烷化工艺技术的开发。针对加压且高放热的耐硫直接甲烷化体系,如何及时有效地移除并充分利用热量是该体系的工艺技术开发的一大挑战。流化床是适宜的耐硫甲烷化反应器。如何降低流化床中催化剂的磨损和流失、避免局部过热造成催化剂烧结是重要的研究方向。与此同时,催化剂也应具备相应的宽活性温度范围、高机械强度和良好的抗烧结性能。
[1]Kopyscinski J, Schildhauer T J, Biollaz S M A.Production of synthetic natural gas (SNG) from coal and dry biomass – a technology review from 1950 to 2009 [J].Fuel, 2010, 89(8): 1763-1783.
[2]Yan X, Liu Y, Zhao B, Wang Z, Wang Y, Liu C J.Methanation over Ni/SiO2: effect of the catalyst preparation methodologies [J].International Journal of Hydrogen Energy, 2013, 38(5): 2283-2291.
[3]Zhao A, Ying W, Zhang H, Ma H, Fang D.Ni-Al2O3catalysts prepared by solution combustion method for syngas methanation [J].Catalysis Communications, 2012, 17: 34-38.
[4]Liu Z, Chu B, Zhai X, Jin Y, Cheng Y.Total methanation of syngas to synthetic natural gas over Ni catalyst in a micro-channel reactor [J].Fuel, 2012, 95: 599-605.
[5]Hu D, Gao J, Ping Y, Jia L, Gunawan P, Zhong Z, Xu G, Gu F, Su F.Enhanced investigation of CO methanation over Ni/Al2O3catalysts for synthetic natural gas production [J].Industrial and Engineering Chemistry Research, 2012, 51(13): 4875-4886.
[6]Wang B, Liu S, Hu Z, Li Z, Ma X.Active phase of highly active Co3O4catalyst for synthetic natural gas production [J].RSC Advances, 2014, 4(100): 57185-57191.
[7]Zhu H, Razzaq R, Jiang L, Li C.Low-temperature methanation of CO in coke oven gas using single nanosized Co3O4catalysts [J].Catalysis Communications, 2012, 23: 43-47.
[8]Eckle S, Augustin M, Anfang H G, Behm R J.Influence of the catalyst loading on the activity and the CO selectivity of supported Ru catalysts in the selective methanation of CO in CO2containing feed gases [J].Catalysis Today, 2012, 181(1): 40-51.
[9]Galletti C, Specchia S, Specchia V.CO selective methanation in H2-rich gas for fuel cell application: microchannel reactor performance with Ru-based catalysts [J].Chemical Engineering Journal, 2011, 167(2/3): 616-621.
[10]Gao J, Liu Q, Gu F, Liu B, Zhong Z, Su F.Recent advances in methanation catalysts for the production of synthetic natural gas [J]. RSC Advances, 2015, 5(29): 22759-22776.
[11]Happel J, Hnatow M A, Bajars L.Methods of making high activity transition metal catalysts[P]: US, 4491639.1985-01-01.
[12]Alex M G, Steffgen F W.Catalytic methanation [J].Catalysis Reviews, 1974, 8(1): 159-210.
[13]Logadóttir A, Moses P G, Hinnemann B, Topsøe N Y, Knudsen K G, Topsøe H, Nørskov J K.A density functional study of inhibition of the HDS hydrogenation pathway by pyridine, benzene, and H2S on MoS2-based catalysts [J].Catalysis Today, 2006, 111(1/2): 44-51.
[14]Chianelli R R, Berhault G, Raybaud P, Kasztelan S, Hafner J,Toulhoat H.Periodic trends in hydrodesulfurization: in support of the Sabatier principle [J].Applied Catalysis A: General, 2002, 227(1/2):83-96.
[15]Alonso G, Berhault G, Aguilar A, Collins V, Ornelas C, Fuentes S,Chianelli R R.Characterization and HDS activity of mesoporous MoS2catalysts prepared by in situ activation of tetraalkylammonium thiomolybdates [J].Journal of Catalysis, 2002, 208(2): 359-369.
[16]Okamoto Y, Maezawa A, Imanaka T.Active sites of molybdenum sulfide catalysts supported on Al2O3and TiO2for hydrodesulfurization and hydrogenation [J].Journal of Catalysis,1989, 120(1): 29-45.
[17]Bui V N, Laurenti D, Afanasiev P, Geantet C.Hydrodeoxygenation of guaiacol with CoMo catalysts (Ⅰ): Promoting effect of cobalt on HDO selectivity and activity [J].Applied Catalysis B: Environmental, 2011, 101(3/4): 239-245.
[18]Badawi M, Paul J F, Cristol S, Payen E, Romero Y, Richard F, Brunet S, Lambert D, Portier X, Popov A, Kondratieva E, Goupil J M, El Fallah J, Gilson J P, Mariey L, Travert A, Maugé F.Effect of water on the stability of Mo and CoMo hydrodeoxygenation catalysts: a combined experimental and DFT study [J].Journal of Catalysis, 2011, 282(1): 155-164.
[19]Christensen J M, Jensen P A, Jensen A D.Effects of feed composition and feed impurities in the catalytic conversion of syngas to higher alcohols over alkali-promoted cobalt-molybdenum sulfide [J].Industrial and Engineering Chemistry Research, 2011, 50(13): 7949-7963.
[20]Gao J, Wang Y, Ping Y, Hu D, Xu G, Gu F, Su F.A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas [J].RSC Advances, 2012, 2(6): 2358-2368.
[21]Lin Hualin(蔺华林), Li Kejian(李克健), Zhao Lijun(赵利军).Research progress of coal-based high temperature methanationcatalyst for synthetic natural gas [J].Chemical Industry and Engineering Progress(化工进展), 2011, 30(8): 1739-1743.
[22]Hu Dacheng(胡大成), Gao Jiajian(高加俭), Jia Chunmiao(贾春苗), Ping Yuan(平原), Jia Lihua(贾丽华), Wang Yingli(王莹利), Xu Guangwen(许光文), Gu Fangna(古芳娜), Su Fabing(苏发兵).Research advances in methanation catalysts and their catalytic mechanisms [J].The Chinese Journal of Process Engineering(过程工程学报), 2011, 11(05): 880-893.
[23]Zhang Cheng(张成).Research progress of methanation of carbon monoxide and carbon dioxide [J].Chemical Industry and Engineering Progress(化工进展), 2007, 26(9): 1269-1273.
[24]Wang W, Wang S, Ma X, Gong J.Recent advances in catalytic hydrogenation of carbon dioxide [J].Chemical Society Reviews, 2011, 40(7): 3703-3727.
[25]Wang W, Gong J.Methanation of carbon dioxide: an overview [J].Frontiers of Chemical Science and Engineering, 2011, 5(1): 2-10.
[26]Cui Kaikai(崔凯凯), Zhou Guilin(周桂林), Xie Hongmei(谢红梅).Research progress in CO2methanation catalysts [J].Chemical Industry and Engineering Progress(化工进展), 2015, 34(3): 724-730,737.
[27]Vasudevan P T, Zhang F.Characterization of supported molybdenum sulfide catalyst ex ammonium tetrathiomolybdate [J].Applied Catalysis A: General, 1994, 112(2): 161-173.
[28]Wang H, Li Z, Wang B, Ma X, Qin S, Sun S, Sun Q.Precursor effect on catalytic properties of Mo-based catalyst for sulfur-resistant methanation [J].Korean Journal of Chemical Engineering, 2014, 31(12): 2157-2161.
[29]Kim M Y, Ha S B, Koh D J, Byun C, Park E D.CO methanation over supported Mo catalysts in the presence of H2S [J].Catalysis Communications, 2013, 35: 68-71.
[30]Fu Yilu(伏义路), Lu Weijie(陆炜杰), Huang Zhigang(黄志刚), Jiang Jiale(姜家乐).Study of methanation and O2chemisorption with several supported sulfide molybdenum catalysts [J].Journal of China University of Science and Technology(中国科学技术大学学报), 1989, 16(2): 171-177.
[31]Qin Shaodong(秦绍东), Long Junying(龙俊英), Tian Dayong(田大勇), Wang Guogao(汪国高), Yang Xia (杨霞), Sun Shouli(孙守理), Sun Qi(孙琦).Supported Mo-based catalysts with different carriers for methanation [J].Industrial Catalysis(工业催化), 2014, 22(10): 770-774.
[32]Wang H, Li Z, Wang E, Lin C, Shang Y, Ding G, Ma X, Qin S, Sun Q.Effect of composite supports on the methanation activity of Co-Mo-based sulphur-resistant catalysts [J].Journal of Natural Gas Chemistry, 2012, 21(6): 767-773.
[33]Mestl G, Srinivasan T K K.Raman spectroscopy of monolayer-type catalysts: supported molybdenum oxides [J].Catalysis Reviews: Science and Engineering, 1998, 40(4): 451-570.
[34]Wachs I E.Raman and IR studies of surface metal oxide species on oxide supports: supported metal oxide catalysts [J].Catalysis Today, 1996, 27(3/4): 437-455.
[35]Wang B, Ding G, Shang Y, Lv J, Wang H, Wang E, Li Z, Ma X, Qin S, Sun Q.Effects of MoO3loading and calcination temperature on the activity of the sulphur-resistant methanation catalyst MoO3/γ-Al2O3[J].Applied Catalysis A: General, 2012, 431/432: 144-150.
[36]Li C P, Hercules D M.A surface spectroscopic study of sulfided molybdena-alumina catalysts [J].Journal of Physical Chemistry, 1984, 88(3): 456-464.
[37]Xu Xianping(徐献平), Zhao Biying(赵璧英), Xie Youchang(谢有畅), Tang Youqi(唐有祺), Yang Xianchun(杨先春).Study on the states of molybdena in MoO3/γ-Al2O3and their formation conditions [J].Chinese Journal of Catalysis(催化学报), 1992, 13(2): 97-102.
[38]Sakashita Y.Effects of surface orientation and crystallinity of alumina supports on the microstructures of molybdenum oxides and sulfides [J].Surface Science, 2001, 489(1/2/3): 45-58.
[39]Sakashita Y, Araki Y, Shimada H.Effects of surface orientation of alumina supports on the catalytic functionality of molybdenum sulfide catalysts [J].Applied Catalysis A: General, 2001, 215(1/2): 101-110.
[40]Yao Yuqin(姚玉芹), Liu Sihan(刘思含), Hu Zongyuan(胡宗元), Wang Baowei(王保伟).Effects of alumina properties on the activity of Mo-based catalyst for sulfur-resistant methanation [J].Petrochemical Technology(石油化工), 2014, 43(7): 754-758.
[41]Chen J, Maugé F, El Fallah J, Oliviero L.IR spectroscopy evidence of MoS2morphology change by citric acid addition on MoS2/Al2O3catalysts —a step forward to differentiate the reactivity of M-edge and S-edge [J].Journal of Catalysis, 2014, 320(1): 170-179.
[42]Li Z, Tian Y, He J, Wang B, Ma X.High CO methanation activity on zirconia-supported molybdenum sulfide catalyst [J].Journal of Energy Chemistry, 2014, 23(5): 625-632.
[43]El-Sharkawy E A, Khder A S, Ahmed A I.Structural characterization and catalytic activity of molybdenum oxide supported zirconia catalysts [J].Microporous and Mesoporous Materials, 2007, 102(1/2/3): 128-137.
[44]Zhang Shenghong( 张 胜 红), Zhang Hongpeng( 张 鸿 鹏), Li Weizhen(李为臻), Zhang Wei(张伟), Huang Hua(黄华), Liu Haichao(刘海超).Effects of zirconia crystallite phases on the structures of MoOx/ZrO2catalysts and their properties in the selective oxidation of methanol [J].Acta Phys.Chim.Sin.(物理化学学报), 2010, 26(7): 1879-1886.
[45]Yamasaki M, Habazaki H, Asami K, Izumiya K, Hashimoto K.Effect of tetragonal ZrO2on the catalytic activity of Ni/ZrO2catalyst prepared from amorphous Ni-Zr alloys [J].Catalysis Communications, 2006, 7(1): 24-28.
[46]Rhodes M D, Bell A T.The effects of zirconia morphology on methanol synthesis from CO and H2over Cu/ZrO2catalysts(Ⅰ): Steady-state studies [J].Journal of Catalysis, 2005, 233(1): 198-209.
[47]He D, Ding Y, Luo H, Li C.Effects of zirconia phase on the synthesis of higher alcohols over zirconia and modified zirconia [J].Journal of Molecular Catalysis A: Chemical, 2004, 208(1/2): 267-271.
[48]Yao H C, Yao Y F Y.Ceria in automotive exhaust catalysts(Ⅰ): Oxygen storage [J].Journal of Catalysis, 1984, 86(2): 254-265.
[49]Tada S, Shimizu T, Kameyama H, Haneda T, Kikuchi R.Ni/CeO2catalysts with high CO2methanation activity and high CH4selectivity at low temperatures [J].International Journal of Hydrogen Energy, 2012, 37(7): 5527-5531.
[50]Zhuang Q, Qin Y, Chang L.Promoting effect of cerium oxide in supported nickel catalyst for hydrocarbon steam-reforming [J].Applied Catalysis, 1991, 70(1): 1-8.
[51]Xu G, Shi K, Gao Y, Xu H, Wei Y.Studies of reforming natural gas with carbon dioxide to produce synthesis gas(Ⅹ): The role of CeO2and MgO promoters [J].Journal of Molecular Catalysis A: Chemical, 1999, 147(1/2): 47-54.
[52]Wang Minwei(王敏炜), Wei Wenlong(魏文龙), Luo Laitao(罗来涛).Preparation of CeO2and its role as catalyst support [J].Chemical Industry and Engineering Progress(化工进展), 2006, 25(5): 517-519.
[53]Wang B, Shang Y, Ding G, Lv J, Wang H, Wang E, Li Z, Ma X, Qin S, Sun Q.Effect of the ceria-alumina composite support on the Mo-based catalyst’s sulfur-resistant activity for the synthetic natural gas process [J].Reaction Kinetics, Mechanisms and Catalysis, 2012, 106(2): 495-506.
[54]Jiang M, Wang B, Yao Y, Wang H, Li Z, Ma X, Qin S, Sun Q.The role of the distribution of Ce species on MoO3/CeO2-Al2O3catalysts in sulfur-resistant methanation [J].Catalysis Communications, 2013, 35: 32-35.
[55]Jiang M, Wang B, Lv J, Wang H, Li Z, Ma X, Qin S, Sun Q.Effect of sulfidation temperature on the catalytic activity of MoO3/CeO2-Al2O3toward sulfur-resistant methanation [J].Applied Catalysis A: General, 2013, 466: 224-232.
[56]Jiang M, Wang B, Yao Y, Li Z, Ma X, Qin S, Sun Q.A comparative study of CeO2-Al2O3supported prepared with different methods and its application on MoO3/CeO2-Al2O3catalyst for sulfur-resistant methanation [J].Applied Surface Science, 2013, 285(PARTB): 267-277.
[57]Sasaki T, Suzuki T.Sulfide molybdenum catalysts for water-gas shift reaction: influence of the kind of promoters and supports to generate MoS2[J].Applied Catalysis A: General, 2014, 484: 79-83.
[58]Chen Y Y, Dong M, Wang J, Jiao H.Mechanisms and energies of water gas shift reaction on Fe-, Co-, and Ni-promoted MoS2catalysts [J].Journal of Physical Chemistry C, 2012, 116(48): 25368-25375.
[59]Qi Xingguo(祁兴国), Dong Qun(董群), Ma Shoubo(马守波), Zhao Fajun(赵法军), Sun Yanping(孙艳萍).Edge structures of molybdenum-based sulfide catalyst [J].Chemical Industry and Engineering Progress(化工进展), 2004, 23(12): 1291-1295.
[60]Jiang M, Wang B, Yao Y, Li Z, Ma X, Qin S, Sun Q.Effect of sulfidation temperature on CoO-MoO3/γ-Al2O3catalyst for sulfur-resistant methanation [J].Catalysis Science and Technology, 2013, 3(10): 2793-2800.
[61]Wang B, Hu Z, Liu S, Jiang M, Yao Y, Li Z, Ma X.Sulphidation temperature on the performance of NiO-MoO3/γ-Al2O3catalysts for sulphur-resistant methanation [J].RSC Advances, 2014, 4(99): 56174-56182.
[62]Li Zhenhua(李振花), Wang Erdong(王二东), Ding Guozhong(丁国忠), Shang Yuguang(尚玉光), Wang Haiyang(王海洋), Wang Baowei(王保伟), Lü Jing(吕静), Ma Xinbin(马新宾), Qin Shaodong(秦绍东).Effect of Mo loading and additive Co on activity of sulfur-resistant methanation catalyst [J].Journal of Tianjin University: Science and Technology(天津大学学报), 2013, 46(6): 546-552.
[63]Lin C, Wang H, Li Z, Wang B, Ma X, Qin S, Sun Q.Effect of a promoter on the methanation activity of a Mo-based sulfur-resistant catalyst [J].Frontiers of Chemical Science and Engineering, 2013, 7(1): 88-94.
[64]Shang Yuguang(尚玉光), Wang Baowei(王保伟), Li Zhenhua(李振花), Ma Xinbin(马新宾), Qin Shaodong(秦绍东), Sun Qi(孙琦).Mo-based catalyst modified with sulfur for sulfur-resistant methanation [J].Petrochemical Technology(石油化工), 2012, 41(9): 999-1004.
[65]Arnoldy P, van den Heijkant J A M, de Bok G D, Moulijn J A.Temperature-programmed sulfiding of MoO3Al2O3catalysts [J].Journal of Catalysis, 1985, 92(1): 35-55.
[66]Farag H.Effect of sulfidation temperatures on the bulk structures of various molybdenum precursors [J].Energy & Fuels, 2002, 16: 944-950.
[67]De Boer M, Van Dillen A J, Koningsberger D C, Geus J W.The structure of highly dispersed SiO2-supported molybdenum oxide catalysts during sulfidation [J].Journal of Physical Chemistry, 1994, 98(32): 7862-7870.
[68]Scheffer B, Arnoldy P, Moulijn J A.Sulfidability and hydrodesulfurization activity of Mo catalysts supported on alumina, silica, and carbon [J].Journal of Catalysis, 1988, 112(2): 516-527.
[69]Weber Th, Muijsers J C, Van Wolput J H M C, Verhagen C P J, Niemantsverdriet J W.Basic reaction steps in the sulfidation of crystalline MoO3to MoS2, as studied by X-ray photoelectron and infrared emission spectroscopy [J].Journal of Physical Chemistry, 1996, 100(33): 14144-14150.
[70]Shi X R, Wang J, Hermann K.Theoretical cluster studies on the catalytic sulfidation of MoO3[J].Journal of Physical Chemistry C, 2010, 114(14): 6791-6801.
[71]Ratnasamy P, Sivasanker S.Structural chemistry of Co-Mo-Alumnina catalysts [J].Catalysis Reviews: Science and Engineering, 1980, 22(3): 401-429.
[72]Topsøe H, Clausen B S, Candia R, Wivel C, Mørup S.In situ Mössbauer emission spectroscopy studies of unsupported and supported sulfided CoMo hydrodesulfurization catalysts: evidence for and nature of a CoMoS phase [J].Journal of Catalysis, 1981, 68(2): 433-452.
[73]Topsøe H, Clausen, B S.Importance of Co-Mo-S type structures in hydrodesulfurization [J].Catalysis reviews, 1984, 26(3/4): 395-420.
[74]Lauritsen J V,Helveg S,Lægsgaard E,Stensgaard I,Clausen B S,Topsøe H,Besenbacher F.Atomic-scale structure of Co-Mo-S nanoclusters in hydrotreating catalysts [J].Journal of Catalysis, 2001, 197(1): 1-5.
[75]Coulier L, De Beer V H J, Van Veen J A R, Niemantsverdriet J W.On the formation of cobalt-molybdenum sulfides in silica-supported hydrotreating model catalysts [J].Topics in Catalysis, 2000, 13(1/2): 99-108.
[76]Van Veen J A R, Gerkema E, Van Der Kraan A M, Knoester A.A real support effect on the activity of fully sulphided CoMoS for the hydrodesulphurization of thiophene [J].Journal of the Chemical Society, Chemical Communications, 1987, 22: 1684-1686.
[77]Bouwens S M A M, Vanzon F B M, Vandijk M P, Vanderkraan A M, Debeer V H J, Vanveen J A R, Koningsberger D C.On the Structural differences between alumina-supported CoMoS Type I and alumina-, silica-, and carbon-supported CoMoS Type II Phases studied by XAFS, MES, and XPS [J].Journal of Catalysis, 1994, 146(2): 375-393.
[78]Al-Zeghayer Y S, Sunderland P, Al-Masry W, Al-Mubaddel F, Ibrahim A A, Bhartiya B K, Jibril B Y.Activity of CoMo/γ-Al2O3as a catalyst in hydrodesulfurization: Effects of Co/Mo ratio and drying condition [J].Applied Catalysis A: General, 2005, 282(1/2): 163-171.
[79]Palcheva R, Spojakina A, Jiratova K, Kaluza L.Effect of Co on HDS activity of alumina-supported heteropolymolybdate [J].Catalysis Letters, 2010, 137(3/4): 216-223.
[80]Wang B, Yao Y, Jiang M, Li Z, Ma X, Qin S, Sun Q.Effect of cobalt and its adding sequence on the catalytic performance of MoO3/Al2O3toward sulfur-resistant methanation [J].Journal of Energy Chemistry, 2014, 23(1): 35-42.
[81]Venezia A M, La Parola V, Deganello G, Cauzzi D, Leonardi G, Predieri G.Influence of the preparation method on the thiophene HDS activity of silica supported CoMo catalysts [J].Applied Catalysis A: General, 2002, 229(1/2): 261-271.
[82]Kempegowda R S, Laosiripojana N, Assabumrungrat S.High temperature desulfurization over nano-scale high surface area ceria for application in SOFC [J].Korean Journal of Chemical Engineering, 2008, 25(2): 223-230.
[83]Jiang M, Wang B, Yao Y, Wang H, Li Z, Ma X, Qin S, Sun Q.Effect of stepwise sulfidation on a MoO3/CeO2-Al2O3catalyst for sulfur-resistant methanation [J].Applied Catalysis A: General, 2014, 469: 89-97.
[84]Fan Chongzheng(范崇正), Sun Hanfang(孙汉芳), Huang Xinnan(黄新楠), Jiang Zhanchang(江展昌).The preparation and activity determination of KSM-01 sulfided molybdenum catalyst for CO methanation reaction [J].Journal of Fuel Chemistry and Technology(燃料化学学报), 1988, 16(2): 188-192.
[85]Shi X R, Jiao H, Hermann K, Wang J.CO hydrogenation reaction on sulfided molybdenum catalysts [J].Journal of Molecular Catalysis A: Chemical, 2009, 312(1/2): 7-17.
