利用DNA洗牌技术一次性克隆多个基因
2015-07-07王肖猛孟祥潮曹雪松
王肖猛,孟祥潮,曹雪松
(聊城大学 生命科学学院,山东 聊城 252300)
利用DNA洗牌技术一次性克隆多个基因
王肖猛,孟祥潮,曹雪松
(聊城大学 生命科学学院,山东 聊城 252300)
目的 探讨DNA洗牌技术(DNA shuffling)对蛋白质组学和代谢组学等研究的意义。方法 利用DNA洗牌技术一次性克隆多个基因片段到表达载体,并同时突变基因内部的2个氨基酸位点,PCR、酶切、测序检测构建表达载体的准确性。结果 成功地一次性克隆了3个编码小RNA的串联短片段MSb1、MSb2、MSb3以及3个蛋白编码基因FokI、MS2、FokI,并一次性将PLRV-P0基因内部2个编码色氨酸(W)的密码子TGG突变为编码丙氨酸(A)的GCG密码子。结论 DNA洗牌技术可应用于基因组学、多基因功能鉴定、融合基因表达和一次性构建基因内部多个点突变的研究,具有精确、快速和高效的特点。
DNA洗牌技术;基因;克隆
随着人类及越来越多生物的全基因组测序的完成,功能基因组研究需要快速和高通量基因克隆技术以用于研究代谢组学、蛋白质组学等,甚至于合成含有多个基因的新组合生物体系[1-6]。近年来开发了一些新技术以用于多片段、高通量DNA的克隆,已得到商业化推广的高通量克隆技术是位点特异性重组克隆系统(site-specific recombinational cloning systems),包括GATEWAY系统[7-9]和In-fusion系统[10-12]。虽然这些技术已被广泛使用,但依然存在以下缺陷:①2种系统都需要将目的基因先通过重组反应克隆到入门载体(entry vector,gateway system)或控制载体(master vector,in-fusion system),再通过一步重组反应将基因克隆到目的表达载体,因此操作步骤繁琐;②由于需要二步反应,加之参与反应的酶不止一种,因而费用较高;③在目的载体中基因两侧或一侧会产生多余的碱基(Gateway技术在基因两侧产生25个,In-fusion技术在基因上游产生34个碱基)。当需要克隆多个串联基因或在基因的两端加上标签时会产生多余的连接氨基酸(分别为8和11个);④需要特定的载体系统。所用载体都含有重组反应所需的特定序列,因而载体需经过改造,并且不能与现有载体实现共享。
DNA“洗牌术”又称DNA改组技术[13-18],或DNA体外随机拼接技术,它的原意是指将来源不同但功能相同的一组同源基因,用核酸酶Ⅰ消化成随机片段,由这些随机片段组成一个文库,使之互为引物和模板进行PCR扩增,当一个基因拷贝片段作为另一基因拷贝的引物时,引起模板互换,重组因而发生。DNA洗牌术为基因克隆和载体构建技术提供了新思路,从而达到方便、快捷和节约克隆基因的目的。本文旨在通过PCR,结合DNA洗牌技术原理和传统酶切-连接克隆技术,分别将3个基因片段及3个小片段DNA串联克隆入表达载体。采用一步法定点突变基因内部2个氨基酸位点,以期省略传统克隆所需的分步酶切-连接的繁琐步骤,提高多基因(或DNA片段)和产生多位点突变基因的克隆效率。
1 材料与方法
1.1 材料
1.1.1 质粒和菌株:载体质粒pCAMBIA1305和pUC-24MS2以及pGR107来自本实验室,pST1374购于Addgene公司,大肠杆菌E.coli DH5α菌株为本实验室保存,感受态为本实验室自制。
1.1.2 工具酶和试剂:质粒小量抽提试剂盒和DNA胶回收试剂盒(AXYGEN)购于Promega公司,EcoR Ι等各种限制性内切酶、PrimeSTAR HS DNA Polymerase with GC Buffer、卡那霉素、氨苄霉素以及T4 DNA连接酶均购于Takara公司。
1.1.3 合成和测序:引物合成在北京三博远志生物技术有限责任公司,测序在上海生工生物工程技术服务有限公司。
1.2 方法
1.2.1 一次性克隆三个串联短片段DNA:首先用传统构建载体的方法在多克隆位点中插入Polymerase III类的启动子AtU6,可以启动小RNA基因的表达。该载体命名为p1305-U6,利用该载体中U6启动子下游的Kpn I和Hind III二个酶切位点,通过DNA 洗牌方法一次性克隆3个与MS2噬菌体外壳蛋白结合的小RNA基因片段(MS binding,MSb)。根据文献报道合成MSb碱基序列,设计并合成引物见表1。

表1 合成MS 结合的PCR引物Tab.1 PCR primers for MS binding gene
将化学合成的6条寡聚核苷酸链(5’端磷酸化修饰)分为MSb1F和MSb1R、MSb2F和MSb2R、MSb3F和MSb3R 3组,每组2条寡聚核苷酸均溶于TE缓冲液,然后每组2条寡聚核苷酸以等摩尔混合,分别经高温变性、退火形成互补双链,其中每两组之间的3’端和5’端分别含有互补的粘性末端,以形成按顺序衔接、相互串联的3个DNA片段。
1.2.2 一次性克隆3个编码蛋白基因:通过DNA 洗牌方法一次性克隆3个长片段串联基因,即形成FokI:MS2:FokI。设计并合成引物见表2。
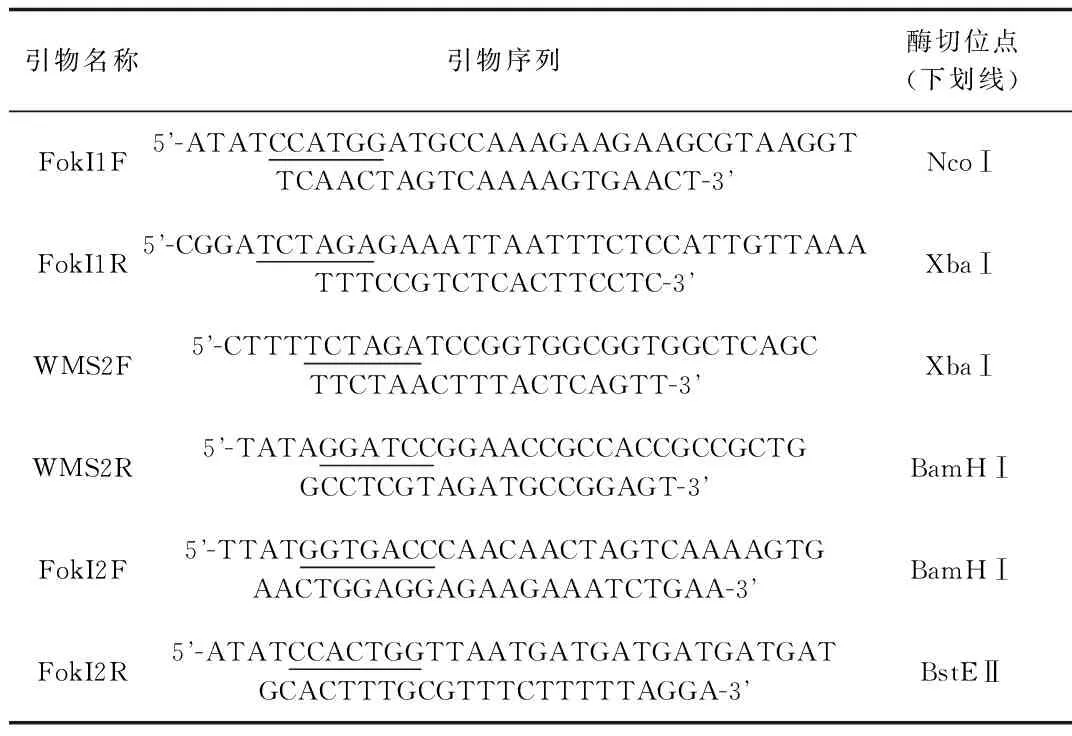
表2 FokI和MS2基因的PCR引物Tab.2 PCR primers for FokI and MS2 genes
以pST1374为模板,FokI1F和FokI1R为引物,进行PCR扩增,其两端分别引入Nco I和Xba I酶切位点。琼脂糖凝胶电泳检测扩增产物,进一步用回收试剂盒回收目的产物,得到目的片段FokI。以质粒pUC-24MS2为模板,WMS2F和WMS2R为引物,采用上步方法获得目的片段MS2,两端分别引入限制性酶切位点XbaⅠ和BamHⅠ。同样方法得到第2个FokI片段,其两端引入酶切位点BamHⅠ和BstEⅡ,这样得到3组双链DNA,用相对应的限制性内切酶酶切并胶回收上述3个PCR片段,每2组之间的3’端和5’端都含有互补的粘性末端,形成按顺序衔接、相互串联的3个融合基因。
1.2.3 一次性克隆并突变基因内部的2个氨基酸位点:PLRV-P0 能够和宿主AGO 1蛋白发生相互作用,其序列中2个重复的WG 基序是其与AGO1蛋白相互作用的关键氨基酸。通过DNA 洗牌方法,一次性将PLRV-P0基因序列中第 87 位和第 140 位的编码色氨酸(W)的密码子TGG突变为编码丙氨酸(A)的GCG,并克隆到植物表达载体pGR107。质粒pGR107中含有酶切位点ClaⅠ和SalⅠ,根据PLRV-P0的基因序列,设计合成含有相关酶切位点的引物,见表3。

表3 突变2个氨基酸位点的PCR引物Tab.3 PCR primers for two amino acid sites mutation
将合成的6条引物分为P0F和P0WA87R、P0WA87F和P0WA140R、P0WA140F和P0R 3组,分别命名为PW1、PW2、PW3,每组2条引物以等摩尔混合。以pER8(含PLRV的基因组)为模板,分别用3对引物进行PCR扩增。其中PW1 、PW2、PW3的两端分别引入限制性酶切位点Cla Ⅰ和BspQ Ⅰ、BspQ Ⅰ、BspQ Ⅰ和Sal Ⅰ,经相应的双酶切并切胶回收以后3’端和5’端分别含有互补的粘性末端,以形成按顺序相互串联的3个DNA片段。
1.3 退火及PCR程序 退火反应程序为:95 ℃预变性5 min,94 ℃变性30s,60 ℃ 退火30s,72 ℃延伸30s进行30个循环,最后72 ℃延伸5 min。 PCR反应体系50 μL,含Pfu DNA聚合酶0.1 μL,10×PCR buffer 5 μL(含有Mg2+),10mmol/L dNTP 1 μL,模板DNA 1 μL(200 μg/的质粒),正反方向引物(稀释后浓度为10 μM)各2 μL,DNA聚合酶0.1 μL。反应条件为:95 ℃预变性5 min,94 ℃变性30s,55 ℃ 退火30 s,72 ℃延伸30 s, 30个循环,最后72 ℃延伸5 min。
1.4 重组体构建方法 用相应限制性内切酶双酶切载体与目的片段一起等摩尔混合,经T4 DNA连接酶在16 ℃下过夜连接。将连接产物转化大肠杆菌感受态细胞DH5α,涂布于含相应抗生素的LB培养基平板上,37 ℃倒置培养12~16 h,挑取单菌落进行菌落PCR鉴定,小量提取阳性克隆质粒,再进行质粒PCR、酶切鉴定和DNA测序,测序后与目的基因序列比对正确者为重组体。
2 结果
2.1 重组体p1305-U6-3XMSb质粒构建 EcoR I和Hind III双酶切鉴定重组体质粒,经测序可知载体构建成功,构建示意图及结果见图1。
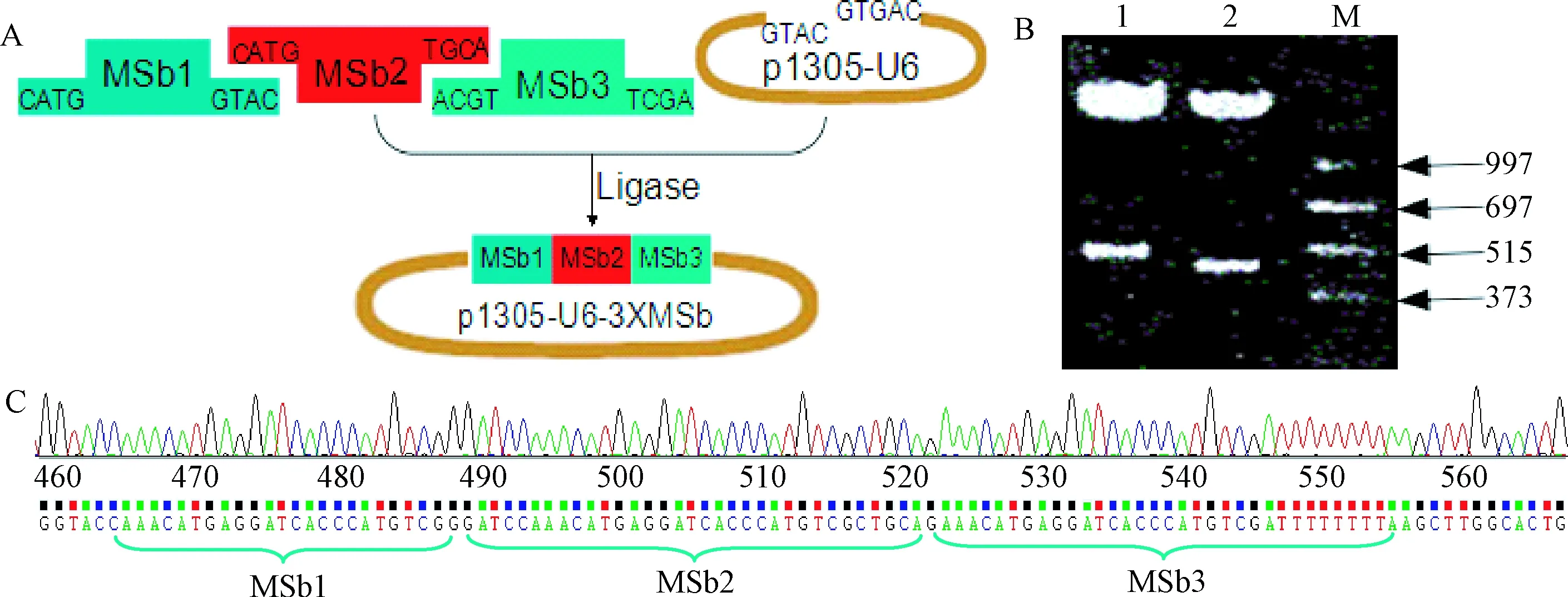
图1 重组体p1305-U6-3XMSb构建图A:重组体p1305-U6-3XMSb的构建示意图。蓝、红、绿色3个矩形分别代表MSb1、MSb2、MSb3 3个短基因片段,突出部分为粘性末端,棕色椭圆为载体;B:重组体经酶切鉴定的电泳结果图。 M.DNA Marker,1.重组质粒用EcoR I和Hind III双酶切得到600bp片段,2.质粒p1305-U6双酶切得到的450bp U6启动子片段;C:3个串联MSb测序峰图,分别用3个括号标明Fig.1 Diagram of p1305-U6-3XMSb constructingA.Diagram of constructing p1305-U6-3XMSb, in which the blue, red and green rectangles represent MSb1, MSb2 and MSb3 segments respectively.Highlights is the sticky end.Brown elliptic is the plasmid; B.Agarose gel electrophoresis results of enzyme digestion for p1305-U6-3XMSb, in which M is DNA Marker,1 column is a recombinant plasmid cleaved by EcoRI and HindIII and 600bp segment, 2 column is the fragment of U6 promoter of 450 bp; C.The diagram of DNA sequencing for 3XMSb
2.2 重组体p1305-FokI:MS2:FokI的构建 以质粒pST1374为模板,PCR扩增目的基因FokI和FokI2,以质粒pUC-24MS2为模板,WMS2F和WMS2R为引物,扩增目的片段MS2。双酶切鉴定重组子,经测序验证载体构建成功,构建过程及结果见图2。
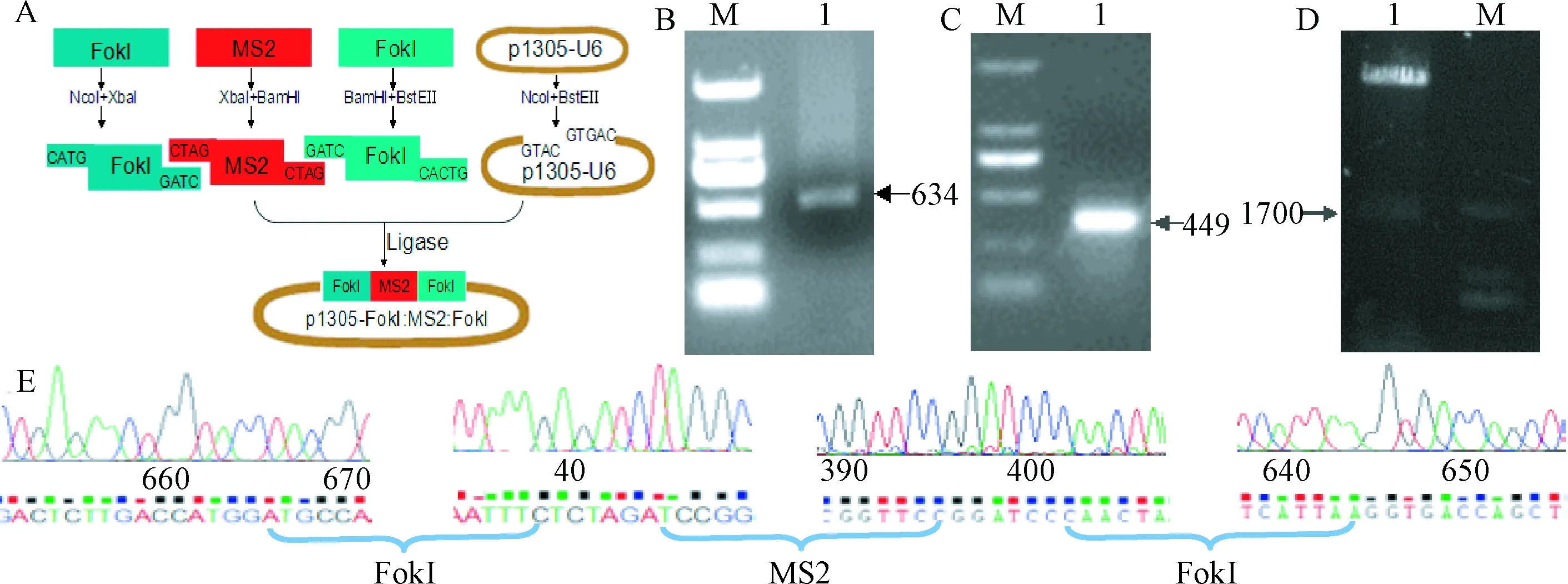
图2 重组体p1305-FokI:MS2:FokI构建图A:重组体p1305-FokI:MS2:FokI的构建示意图。蓝、红、绿色3个矩形分别代表FokI、MS2、FokI 3个编码蛋白的基因片段,突出部分为粘性末端,棕色的椭圆代表载体;B、C:FokI和MS2基因的PCR扩增电泳结果图。M.DNA Marker;1.基因片段,箭头代表片段大小;D:重组体经酶切鉴定的电泳结果图。1.重组质粒经NcoI和BstEII双酶切得到1700bp的含3个蛋白基因的片段;E:3个串联蛋白的测序峰图,分别用大括号标明,因序列太长,中间用省略号代替Fig.2 Diagram of p1305-FokI:MS2:FokI constructingA:Diagram of constructing p1305:FokI:MS2:FokI, in which the blue, red and green rectangles represent FokI , MS2 and FokI segments respectively who encoded protein.Highlights is the sticky end.Brown elliptic is the plasmid;B,C:Agarose gel electrophoresis results of PCR amplification for FokI and MS2.M is DNA Marker; 1 is gene fragments, arrows mean the fragment size;D:Agarose gel electrophoresis of enzyme digestion for p1305-FokI-MS2-FokI, 1 is a recombinant plasmid cleaved by NcoI and BstEII and 1700bp segment linked to FokI, MS2 and FokI; E.The diagram of DNA sequencing for FokI, MS2 and FokI, three tandem genes are depicted.Because the sequence is too long, use ellipses instead of in the middle
2.3 重组体pGR107-P0WA的构建 以pER8的cDNA为模板,分别以PLRV-P0F和POWA87R、P0WA87F和P0WA140R、P0WA140F和PLRV-P0R、PLRV-P0F和PLRV-P0R为引物,PCR扩增得大小约为283 bp、188 bp、370 bp 3个目的片段。用ClaⅠ和SalⅠ双酶切鉴定重组子pGR107-P0WA,得到750 bp目的片段。经测序并序列比对,一次性成功将P0蛋白序列中第87位和第140位的色氨酸(W)点突变为丙氨酸(A),重组体的构建示意图及结果见图3。
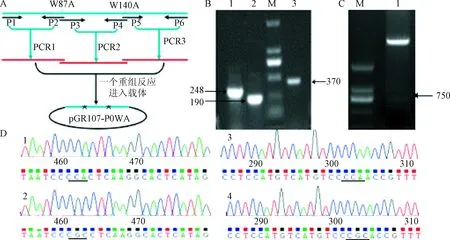
图3 重组体pGR107-P0WA构建图A:重组体pGR107-P0WA的构建示意图。黑色横向箭头为PCR引物;红线为PCR片段;椭圆为质粒,*为突变氨基酸位点;B:PCR扩增的电泳结果图。M.DNA Marker;1,2,3.基因片段,箭头代表片段大小;C:重组体经酶切鉴定的电泳结果图。M.DNA Markev; 1.重组质粒经CalI和SalI双酶切得到的750bp片段;D:突变氨基酸位点的测序峰图。1,2.PLRV-P0基因未突变前序列及峰图,1划横线的位置是第87位色氨酸(W),2是140位色氨酸位置;3、4.基因突变后序列及峰图,划横线的位置分别是突变后的第87位和140位丙氨酸(A)。测序序列为反向互补链Fig.3 Diagram of pGR107-P0WA constructingA:Diagram of constructing pGR107-P0WA,in which black horizontal arrows represent PCR primers, red lines are PCR fragments, oval represents the plasmid,* is complementary sticky end;B.Agarose gel electrophoresis of PCR amplification, in which M is DNA Marker.1,2,3 are gene fragments, arrows mean the fragment size.C.Agarose gel electrophoresis results of enzyme digestion for pGR107-P0WA, 1 is a recombinant plasmid cleaved by Cal and SalI and 750bp segment; D.The diagram of DNA sequencing for pGR107-P0WA.1,2 are gene sequences of PLRV-P0 without mutation, in which 1 row horizontal position is 87th of tryptophan (W), and 2 is 140 bit position of tryptophan.3,4 are gene sequences with mutation, in which the row horizontal position is 87 and 140 of alanine (A) after mutation
3 讨论
后基因组时代的功能基因组和蛋白质组学等研究需要克隆、鉴定大量未知基因的功能,近年来发展起来的合成或整合生物学研究也需要同时克隆和表达一组功能相关基因,而ZFN和TALEN等基因组编辑技术也需要串联克隆多个DNA序列结合蛋白。除了利用重组方法克隆基因外,近年来开发了几种多基因组装技术,如Golden Gate、BioBricks等,其中Golden Gate技术是利用Type II类核酸内切酶,利用其酶的识别与切割序列分离的特性,经酶切割后可以形成各片段首-尾相互配对的粘性末端,如载体被改造成含有同一核酸内切酶位点,则可经同一种酶切割后再连接,该方法对于串联克隆较多DNA片段时非常有效[19],但此方法不能兼顾传统载体上的常用Type II类核酸内切酶;BioBricks方法是利用2个同尾核酸内切酶实现有限片段(一次只能串联2个片段)的连接及克隆[20]。
本文结合Golden Gate和BioBricks克隆方法的优点,同时兼顾传统酶切-连接克隆技术和大部分实验室已有的克隆载体,利用DNA洗牌技术,构建了一种简便、高效的多基因克隆方法。实验结果表明,这是一种简便、通用、高效的克隆基因的方法,具有以下优点:
① 设计操作相对简便。传统方法在构建多片段DNA插入的载体时,每插入一个片段就需要一次连接、转化及阳性克隆鉴定,操作步骤繁琐,通常要构建多个中间载体,要经多次连接、转化,增加了重组体鉴定时的工作量,费时又费力。本文结果表明,对于编码FokI、MS2、FokI 3个蛋白基因和MS2 binding RNA小片段DNA,只要引入合适的首尾相连的酶切位点,就可以一次性克隆入目标载体,实现单个重组反应精确连接多个基因片段,将双酶切-连接克隆简化为单个重组反应,便于分子克隆技术标准化,可以实现大规模克隆,而不影响基因的表达,大大节省了时间,提高了工作效率;② 实现基因之间的无缝连接。如果用Type II S类核酸内切酶,利用其酶的识别与切割序列分离的特性,经酶切割后可以形成各片段首-尾相互配对的粘性末端,并且不含内切酶的识别序列,则可以实现基因之间或基因编码蛋白与标签蛋白之间的无缝连接,即最终克隆的基因无外源冗余氨基酸,所表达的蛋白质可以真实反应其性质;③ 可同时定点突变基因内部多个氨基酸位点。传统构建氨基酸替换突变多用位点重组PCR方法,该方法通过设计重叠引物而引入欲突变位点的编码碱基,并且产生一个突变位点需经二轮PCR,克隆、测序验证后,在此基础上再经同样过程产生第2个突变位点,以此类推。过程非常繁琐且耗费时间。本文利用DNA洗牌术,通过设计含有DNA片段间经酶切后的单链突出互补末端的引物,在基因片段内部一次性引入并克隆2个氨基酸替换突变,极大地简化了实验步骤。对于确定蛋白质结构域中的关键氨基酸、蛋白质分子进化等研究提供了一种有效的技术手段。
[1] Halpin,C.Gene stacking in transgenic plants: the challenge for 21st century plant biotechnology[J].Plant Biotechnol,2005,3(12):141-155.
[2] Capell T,Christou P.Progress in plant metabolic engineering[J].Curr Opin Biotechnol,2004,15(6):148-154.
[3] Dafny M,Tzfira T Delivery ofmultiple transgenes to plant cells[J].Plant Physiol,2007,145(354):1118-1128.
[4] Marsischky G,LaBaer J.Many Paths to Many Clones: A Comparative Look at High-Throughput Cloning Methods[J].Genome Res,2004,14(576):2020-2028.
[5] Ellis T,Adie T,Baldwin G.DNA assembly for synthetic biology: from parts to pathways and beyond[J].Integr Bi,2011,3(10):109-118.
[6] Andrianantoandro S,Basu D,Karig K.Synthetic biology: new engineering rules for an emerging discipline[J].Mol Syst Bio,2006,2(8):2006-0028.
[7] Walhout AJ,Temple GF,Brasch MA,et al.GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes[J].Methods Enzymol,2000,328(246):579-592.
[8] Atanassov II,Atanassov II,Etchells JP,et al.A simple,flexible and efficient PCR-fusion/Gateway cloning procedure for gene fusion,site-directed mutagenesis,short sequence insertion and domain deletions and swaps[J].Plant Methods,2009,5(2):14-20.
[9] Esposito D,Garvey LA,Chakiath CS.Gateway cloning for protein expression[J].Methods Mol Biol,2009,498(190):31-54.
[10] Berrow NS,Alderton D,Owens RJ.The precise engineering of expression vectors using high-throughput in-Fusion PCR cloning[J].Methods Mol Biol, 2009,498(235):75-90.
[11] Zhu B,Cai G,Hall O.In-fusion assembly:seamless engineering of multidomain fusion proteins,modular vectors,and mutations[J].Biotechniques,2007,43(3):354-359.
[12] Berrow S,Alderton D,Sainsbury S.A versatile ligation independent cloning method suitable for high-throughput expression screening applications[J].Nucleic Acids Res,2007,35(6):45-50.
[13] Kikuchi M,Ohnishi K,Harayama S.An effective family shuffling method usingsingle-stranded DNA[J].Gene,2000,243(135):133-137.
[14] Lorimer I A,Pastan I.Random recombination of antibody single chain Fv sequences after fragmentation with DNaseI in the presence of Mn2+[J].Nucleic Acids Res,1995,23(13):3067-3068.
[15] Shao Z,Zhao H,Giver L,et al.Random-priming in vitro recombination:an effective tool for directed evolution[J].Nucleic Acids Res,1998,26(3):681-683.
[16] Stemmer WP.DNA shuffling by random fragmentation and reassembly: in vitrorecombination for molecular evolution.Proc[J].Natl Acad Sci,USA,1994,91(37):10747-10751.[17] Stemmer WP.Rapid evolution of a protein in vitro by DNA shuffling[J].Nature,1994,370(245):389-391.
[18] Zhao H,Arnold FH.Optimization of DNA shuffling for high fidelity recombination[J].Nucleic Acids Res,1997,25(7):1307-1308.
[19] Weber E,Gruetzner R,Werner S.etal.Assembly of Designer TAL Effectors by Golden Gate Cloning[J].PLoS ONE, 2011,6(5):1-5.
[20] Knight T,Thomas F,Reshma PS,et al.Engineering BioBrick vectors from BioBrick parts[J].Journal of Biological Engineering,2008,2 (5):1-12.
(编校:吴茜)
One step cloning multiple genes using DNA shuffling
WANG Xiao-meng,MENG Xiang-chao,CAO Xue-song
(College of Life science, Liaocheng University, Liaocheng 252000, China)
ObjectiveTo explore the application of DNA shuffling method in the field of proteomics or metabolomics.MethodsMultiple genes or DNA fragments were cloned into destination vector and mutations of two amino acid substitutions within the gene were obtained though DNA shuffling, and the constructed plasmids were verdified by PCR, enzyme digestion and sequencing methods.ResultsMultiple tandem genes of MSb1, MSb2, MSb3 encoding small RNAs into destination vector were successfully cloned and two TGG encoding tryptophan to GCG encoding alanine within the sequence of PLRV-P0 gene were mutated simultaneously. ConclusionThis approach can be utilized in genomics, multi-gene identification or multi-site mutagenesis and fusion proteins expression, which is precise, cost-effective and high efficacy.
DNA shuffling; gene; clone
国家自然科学基金(31370387);山东省自然科学基金(ZR2011CM045)
王肖猛,女,硕士,研究方向:植物分子生物学,E-mail:973082527@qq.com。
Q81
A
1005-1678(2015)02-0005-05
