表面等离激元与表面非线性光学
2015-07-02刘韡韬王洪庆
刘韡韬 王洪庆
(复旦大学物理系,应用表面物理国家重点实验室,微纳光子结构教育部重点实验室,上海 200433)
二阶非线性光学效应(如光学二次谐波、光学混频等)是一类独特的表面探测技术[1].实验上,人们通常将频率分别为(ω1,ω2)的两束激光交叠在样品表面,以产生并探测这两束光生成的和频信号(ω1+ω2).这一技术的独特之处在于:当样品的块材结构具有中心反演对称性时,样品体相的响应在电偶极矩近似下往往可以忽略不计,和频信号几乎完全源于表面少数几个单层,这意味着该类技术对材料表面的信息具有很高的灵敏度.更重要的是,这是一类纯光学的探测手段,它们可被用于光子所能达到的任何界面.绝大多数传统的表面探测技术使用各类探针粒子(如电子能量损失谱、氦原子散射、扫描隧道显微镜等),其表面灵敏度来自于这些粒子对材料很低的贯穿能力,使得粒子仅能穿透表面附近的材料薄层,因而散射粒子所携带的信息中有较大比例源于材料表面.然而基于同一原因,各类粒子的平均自由程也往往随气压的增大而急剧减小,因而这些表面技术几乎都必须工作在超高真空环境下.这一限制使得很多拥有重要价值的表面体系难以被涉及:如常压,甚至高压下的表面催化反应、液体表面、具有较高挥发性的聚合物表面、固液界面及其他隐藏界面等.与之形成鲜明对比的是,二阶非线性光学技术的表面灵敏度源于对称性原理而非特定粒子的贯穿深度,因而不受超高真空环境的制约,可以被用于研究上述的各类体系,与传统表面技术有着很强的互补性.20世纪80年代,该类技术首先由加州大学伯克利分校的沈元壤教授课题组运用于表面吸附的研究[2,3],并迅速在表面催化等领域的研究中发挥出显著的作用[4].历经多年的发展,目前二阶非线性光学技术已被广泛应用于表面科学中的多个领域,包括表面分子吸附[5-8]、液体的表面与界面[9-13]、高分子聚合物表面[14-17]、固相的表面与界面[18-21]、生物分子体系表面[22-25]、以及表面超快动力学[26-29],等.在这些工作中,表面非线性光学技术使人们得以探测多种原先无法涉及的体系,并进一步获取到大量分子层面的微观信息,包括表面的分子或结构种类、密度、取向、构型以及动力学属性,成为了表面科学中不可或缺的重要手段.
和频振动光谱(SFVS)的基本原理在一些著作中有详细论述[30,31].简单来讲,频率为ω1和ω2的光场在样品表面重合,引起表面分子的极化,如图1(a)是和频产生过程(SFG)的能级跃迁示意图,分子吸收两个光子被激发,被激发的分子往往不稳定很快跃迁回基态,并放出一个新的光子,由于能量守恒,出射的光子能量等于入射两个光子能量之和.
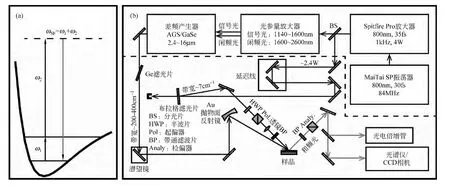
图1 (a)和频产生过程能级跃迁示意图;(b)和频光谱典型装置示意图
和频信号的强度可表述为[32]
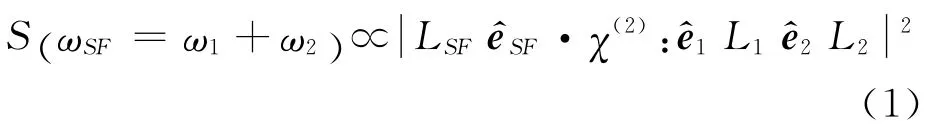
其中χ(2)为表面非线性极化率张量,直接反映了样品表面的信息,如分子面密度、表面取向等,可表述为

在上述两个表达式中,是非共振部分的贡献,Aq,ωq,Γq分别表示第q个振动模式的强度、共振频率和衰减系数.,Li分别对应于频率为ωi的激光的单位极化矢量和菲涅尔系数.对于一个分子数密度为NS、取向分布为f(Ω)的表面,其第q个振动模式的强度Aq可表述为[33]
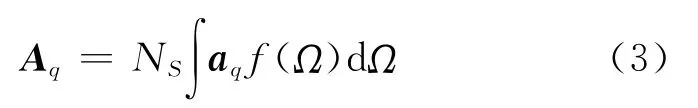
其中aq为单个分子的二阶非线性极化率张量,这里没有考虑局域场的修正.通过不同的偏振组合,可以测量得到χ(2)的各个非零张量元,由χ(2)则可以获得分子尺度上的表面信息.图1(b)显示的是一套典型的和频光谱实验装置,其中虚线以上表示光源部分,由 MaiTai SP振荡器输出一个800nm波长,30fs持续时间,84MHz重频的种子光源,经过Spitfire Pro放大产生1kHz,4W平均功率的高能量飞秒脉冲,激光经过分束,一部分能量再经光参量放大器和差频产生器产生1μm~16μm连续可调的红外飞秒激光;虚线以下表示装置部分,红外和800nm的两路激光经过起偏、聚焦和滤波等操作后重合在样品表面,产生的和频光同样经过滤波和检偏后由光电倍增管和光谱仪等仪器收集探测.
表面等离激元也是一种常见的表面光学现象.在外加电磁场的驱动下,导体内部费米面附近的自由电子可在导体的表面发生集体震荡,产生所谓的表面等离激元.在共振的状态下,电磁场的能量可被有效地转换为导体表面自由电子的集体震荡能.表面等离激元的概念在20世纪初已被提出[34];70年代,Kretschmann与 Otto提出了激发表面等离激元的纯光学手段并被广泛运用[35,36].由于表面等离激元的激发同时往往伴随着表面电场的增强效应,这种现象具有显著的应用价值,因而自20世纪70年代以来得到了极为广泛的研究.一个重要的突破是人们发现,由于金属纳米结构中的限域效应,其等离激元震荡可造成局域电场在远小于光波长尺度上的急剧增强[37],并藉此开发了表面增强拉曼散射[38].90年代纳米科学的发展为这一方向带来了新的契机,1997年人们通过表面增强拉曼散射成功探测到了吸附在单个银纳米颗粒上的单个罗丹明6G染料分子,在不同尺寸、形态的纳米颗粒上观察到了高达1014~1015的拉曼散射增强因子[39].2007年,藉由类似的途径,人们利用相互耦合的银纳米颗粒对,获得了107~1013的增强因子,并实现了对单个Cy5染料分子的表面增强荧光探测[40].这些工作将拉曼光谱、荧光光谱技术带入了单分子领域,实现了过去所难以企及的光谱学灵敏度,也拥有巨大的应用价值.目前,表面等离激元相关的光学、谱学及影像学[41]技术已成为一个极为广阔的研究领域.
表面等离激元可视作是一种在界面上传播的电磁波,图2(a)是在两个半无限大介质1和介质2界面上传播的表面等离激元示意图,其电场可表述为
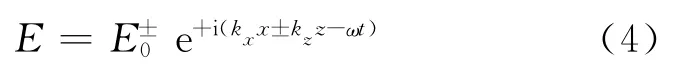
其中+号对应于z≥0,-号对应于z≤0.为了保证表面等离激元的存在,kz必须为纯虚数[31],也就是说在垂直界面的方向上,表面等离激元的电场随距离呈指数衰减

如图2(b)所示.通过麦克斯韦方程组和边界条件,可以推导出表面等离激元的色散关系[42]
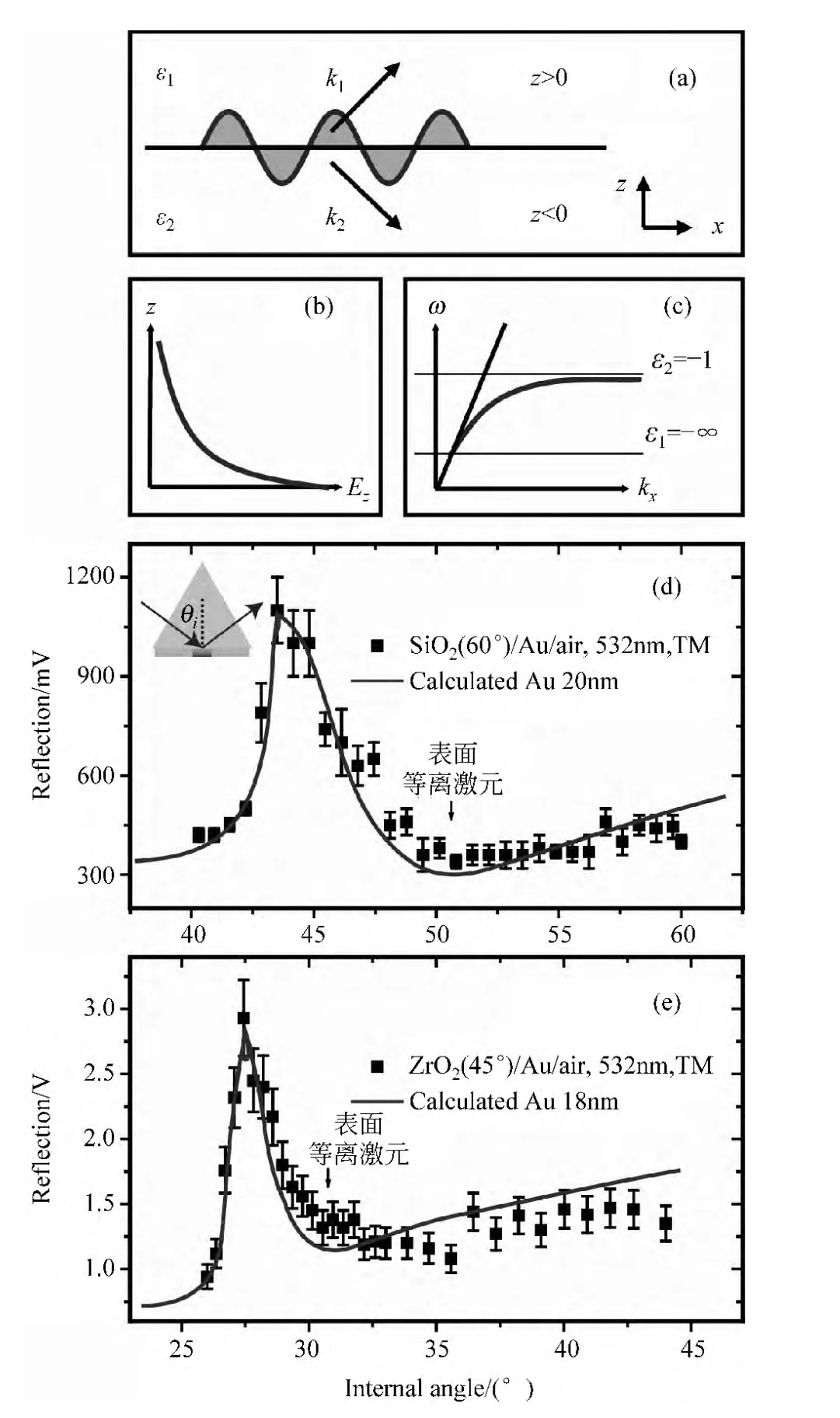
图2 (a)描述表面等离激元在介质1和介质2界面上传播的示意图;(b)在垂直表面方向上,表面等离激元的电场强度与距离的关系;(c)色散关系示意图,粗直线表示光在空气中的色散关系,曲线表示在不同金属表面,等离激元的色散关系变化范围;(d)和(e)利用 Kretschmann配置结构[35]分别在二氧化硅/金界面和二氧化锆/金界面上测量与计算532nm激光反射能量与入射角的依赖关系
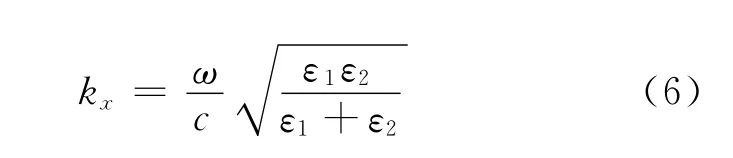
为了更好地说明色散关系的物理意义,这里假设介质1为金属,介质2为空气,如图2(c)所示,粗直线表示光在空气中的色散关系:ω=ckx,曲线表示在不同金属表面,等离激元的色散关系变化范围,由于表面等离激元要求ε1<0且所以这个范围就对应于ε1→-∞ 到ε1=-1,图中细线表示这两个极限,可以看到只有当金属介电常数接近负无穷时,曲线才与粗直线有交点,这表明我们无法直接在空气和金属的界面上激发出表面等离激元.然而,可以参考集成光学中的一些光耦合方法,比如利用棱镜来激发表面等离激元,Otto配置[36]和 Kretschmann配置[35]都是十分有效的方法.如图2(d)和(e),是我们利用Kretschmann配置来激发表面等离激元的两组实验数据,Kretschmann配置的结构示意图显示在(d)左上部分,这是一种表面镀有金属薄膜的棱镜结构,(d)和(e)分别对应二氧化硅和二氧化锆两种材质的棱镜,表面分别镀有20nm和18nm厚度的金膜,波长为532nm的激光透过棱镜入射到金膜表面,当入射角达到相位匹配角时,光就会激发表面等离激元,同时导致反射光能量的减弱,因此测量反射光能量与入射角之间的依赖关系,并与理论计算比较可以帮助我们证明表面等离激元的产生,图中方块表示实验数值,曲线表示理论计算,可以看出两者符合很好,表明在实验中确实激发了表面等离激元.
由于非线性光学过程往往涉及高阶次的电场,可以预见,表面等离激元带来的场增强效应在非线性光学效应中将会更加显著,因而对非线性光学与表面等离激元的结合是相关领域中另一个经久不衰的课题[43].20世纪70年代,沈元壤课题组使用Kretschmann耦合方式进行了表面等离激元结合反斯托克斯拉曼散射的研究,在泵浦光、斯托克斯光与反斯托克斯光频上的表面等离激元达到位相匹配时,反斯托克斯光的信号得到了显著增强,从而探测到了来自于极薄的苯膜的相干反斯托克斯拉曼散射信号[44].自此之后,表面等离激元增强的三阶非线性光学效应获得了广泛研究.人们利用金属纳米颗粒以及金属/介质混合纳米结构中观察到了等离激元对三次谐波显著的增强效应,并借助三次谐波信号探测到了等离激元共振本身的动力学属性[45].目前,人们已可借助等离激元增强探测源于单个纳米颗粒的三次谐波信 号[46].2008年,Novotny课题组同样采取Kretschmann耦合方式,利用四波混频激发了金膜上的表面等离激元[47];之后,同一课题组展示了无需借助特殊耦合方式,即可利用四波混频实现自由空间到金属表面等离激元的位相匹配[48],并在此基础上研制了非线性暗场显微镜,通过表面等离激元衰逝波在微小结构上的散射实现了很高的空间分辨率与信噪比[49].很多研究表明,表面等离激元的场增强效应可使普通材料的三阶非线性光学响应远远超过常用的非线性光学材料[50-53].此外,人们不但利用表面等离激元增强三阶非线性光学效应,在纳米尺度调制、操控三阶非线性光学过程,也同样可以利用这些技术研究等离激元本身的各种属性[54-56].
由于二阶非线性光学过程在中心反演对称的体材料中是电偶极矩禁戒的,与线性甚至三阶非线性光学效应相比,这些源于表面、界面的响应往往对应着非常微弱的信号.与此同时,表面等离激元本身是一种表面模式,也有着对表面成分与结构的灵敏度,故而人们预期,这两种技术的结合能带来更高的表面探测灵敏度以及更广泛的应用范畴.20世纪80年代初,沈元壤课题组已在粗糙的金属银表面观测到了二次谐波信号的急剧增强,并研究了该效应与金属表面电化学反应的相关性[57].近年来,表面等离激元增强的光学二次谐波已获得了大量的研究.人们在不同的纳米结构上观察到了二次谐波信号在等离激元共振频率上的显著增强,包括金属纳米颗粒[58]、金属薄膜上的单个纳米孔洞[59]、周期性的纳米孔阵列[60]以及金属与其他材料的复合纳米结构[61,62]等.2011年,Brongersma课题组在金属电极上刻蚀出周期性结构以激发等离激元共振,并通过同一电极施加电场,观测到外加电场对等离激元共振二次谐波信号强度的调控作用,获得了很高的调控幅度,展示了一种具有实际应用价值的、基于等离激元的非线性光学转换器件[63].在这些研究中,使用贵金属纳米颗粒等造成的二次谐波增强因子可达数千倍,与贵金属纳米颗粒结合的薄膜材料其二次谐波产生效率可与数毫米厚的非线性晶体媲美[64,65].此外,人们还利用二次谐波本身来表征表面等离激元空间结构,获取了纳米结构中表面等离激元“热点”的影像信息[66].除了电偶极矩跃迁之外,多极矩与磁偶极矩跃迁也可产生二次谐波效应,它们相比电偶极矩跃迁所引起的效应往往较弱,故而等离激元共振的场增强效应可为研究这些现象带来很大帮助.Linden等人研究了基于开口谐振环的超构材料中的二次谐波效应及其对材料结构的依赖关系,发现共振增强的二次谐波信号主要源于磁偶极矩跃迁,揭示了超构材料截然不同与常规材料的非线性光学属性[67,68].2012年,Dal Negro等人在金纳米颗粒的平面阵列结构中研究了等离激元共振的二次谐波效应,通过系统化地改变阵列结构的尺寸与周期,证实了该结构对多极矩二次谐波的增强与调制效应.
尽管二次谐波可被视作二阶光学混频的一种特例,表面等离激元与表面光学混频光谱的结合研究却相对而言较为薄弱.原因之一在于表面等离激元是一种共振效应,并且通常只有较窄的共振带宽.当光学混频过程涉及3种不同频率的光波时,通常难以使各个频率都得到增强.对此,很多工作利用了具有一定尺寸分布的贵金属纳米颗粒中的局域等离激元,以实现较宽频率范围内的场增强效应.大多数研究沿用贵金属纳米颗粒,将其置于金属薄膜表面以形成随机或周期性的“热点”,并获取增强的光学混频信号[69-72].2013年,Lis等人系统研究了金属纳米圆柱对表面光学混频光谱的增强效应,通过改变纳米圆柱的密度与径长比,可使混频光谱测量中所采用的可见光以及混频光在不同偏振态上同时得到增强,将表面混频信号提高数十至数百倍,大大提高了对表面吸附硫醇分子的探测灵敏度[73,74].
尽管金属纳米结构有着显著的场增强效应,但表面光学混频技术的一大特点在于对表面分子朝向与构型的解析能力,故而对于平滑表面的研究依然十分重要.在这方面,人们通常利用常见的全反射构型激发表面等离激元共振[75,76],以在平滑表面上获得场增强的效果.由于贵金属的等离子频率往往在可见光区乃至更高频的光学波段,表面等离激元的态密度也在邻近波段内达到最高,人们通常选择在混频光谱测量中所采用的可见光或者混频光的频率上激发表面等离激元.然而,对于一些特定的界面体系,仅仅增强可见光或者混频光的场强并不足以提高光学混频光谱的灵敏度.电化学界面是一个典型的例子.历经数百年的发展,电化学在能源、医药等无数领域发挥着极为重大的作用,然而人们对电化学反应在分子层面上的信息仍知之甚少,包括界面上的分子种类、构型以及反应的瞬态过程等.造成该局面的一个重要原因,即在于电化学界面难以为绝大多数表面分析技术所贯穿,包括常规的表面混频振动光谱:该技术通过红外光获取界面的分子信息,而常用的电极与电解液对红外光都有着强烈的吸收,使介于两者间的电化学界面无法被光子贯穿.为将红外光的能量尽可能耦合至电化学界面,过去的研究往往采用极薄的金属薄膜或极薄的液体层.然而在这些测量过程中,来自不同界面的光学混频信号彼此无法区分,得到的光谱包含的不仅仅是源于电化学界面的信息,还混合着来自于其他界面的无关信号,使结果带有很大的不确定性.在这种情况下,仅仅增强可见光或者混频光的场强显然是难以解决问题的.
为解决这一难题,本文作者与合作者以金属薄膜作为电化学反应的工作电极,通过表面等离激元共振将红外光场高效耦合到电极与电解液的界面处,避免了多个界面共存的问题,可确保测量结果来自于电化学界面本身,同时利用了表面等离激元的场增强效应,大大提高了红外光的耦合效率及相应的表面探测灵敏度.工作中,不但测得了原先难以探测的界面分子光谱,还借助表面等离激元共振本身了解到界面的介电性质,赢得了“1+1>2”的效果[77].图3中演示了表面等离激元激发对金/水界面上光学混频信号显著的增强效应.实验装置图如图3(a)所示:红外光束通过一块熔融石英棱镜入射至棱镜底部电子束蒸镀的20nm厚金膜,利用Kretschmann模式耦合至金膜与水的界面并激发该界面上红外频段的表面等离激元;可见光则通过水入射,与红外表面等离激元叠加产生金/水界面上的光学混频信号.实验中的激光来自于一套10Hz的皮秒激光系统,红外与可见光(532nm)脉冲能量约100~300μJ.为证实表面等离激元对该界面上光学混频信号的增强作用,我们比较了不同实验设置下金表面硫醇自组装分子层的光学混频信号.如图3(b)所示:
曲线代表来自于硫醇自组装分子层与空气界面的混频光谱(红外、可见光皆由空气入射,无表面等离激元激发),背景来自于金的非共振混频信号,位于~2880、2950cm-1处的“凹陷”则源于硫醇分子碳氢键的伸缩振动模式,分别为CH3基团的对称伸缩振动模式、及其与弯曲振动模式间的费米共振[78]; 与 曲线分别代表来自于金/水与金/重水界面的光谱,由于水与重水在该红外波段的折射率不同,红外表面等离激元仅在金/重水界面上得以激发,显然,金/重水界面的光谱强度远大于金/水界面,且源于界面处的硫醇自组装分子层的伸缩振动模式清晰可见,证明了表面等离激元对光学混频信号的显著增强效应.在图3(c)中,定量计算了红外表面等离激元对金/重水界面局域电场的增强效应(实线);如我们所预期的那样,在扣除该部分效应后,金/重水界面硫醇自组装分子层的光谱(虚线)与其在金/空气表面吻合甚好.
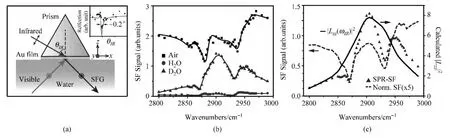
图3 (a)实验装置示意图;(b)金表面硫醇自组装分子层在有/无红外表面等离激元激发下的光学混频光谱;(c)红外表面等离激元增强的局域电场对混频光谱的影响(摘自文献 [77])
金电极在水溶液中的氧化还原是电化学中被广为研究的原型体系之一,然而由于分子级探测手段的缺乏,该反应在微观尺度上的许多机制尚不清楚[79,80].图4(a)展示了纯金工作电极在pH=12氢氧化钾重水溶液中的循环伏安曲线.与此同时,在同一电极上原位获取的表面混频光谱也表现出随工作电极偏压的剧烈变化[图4(b)].由于光谱的峰值频率变化较小,我们将红外频率固定在2865cm-1,而记录光学混频信号随工作电极偏压的变化[图4(c)].对比图4(a)与(c),可以看到光学混频信号与循环伏安曲线明确的对应关系.将氧化过程(负偏压扫至正偏压,向右箭头所示曲线)分为3个区域:Ⅰa,Ⅱa与Ⅲa.Ⅰa区域对应的电化学过程是金电极表面双电层的电荷聚集过程,伴随着界面静电场的增强及其导致的光学混频信号的增强.Ⅱa区域中+1.4V的峰值电流对应金表面的氧化过程,这一过程中具体的生成物在实验上始终有所争议[80,81].在我们的实验中,由于光学混频信号在这一区域持续增强,意味着生成物应以极性且有序的界面AuOD基团为主,而不是弱极性且无序的Au2O3等结构.Ⅲa区域中,金的表面进一步氧化,循环伏安曲线的电流持续增加,而光学混频信号则急剧下降,意味着这一阶段的生成物应以弱极性且无序的Au2O3结构为主,并与文献中的“place-exchange”过程相对应[79].还原过程(正偏压扫至负偏压,向左箭头所示曲线)也可分为3个区域:Ⅰc,Ⅱc与Ⅲc.与氧化过程一样,各个还原峰所对应的微观机制也存有争议[79,80].结合光谱信息,我们发现Ⅰc还原过程中光学混频信号迅速增强,并在这一过程结束时达到Ⅲa氧化过程开始时的强度,表示Ⅰc过程中被还原的界面结构也即Ⅲa过程中、以Au2O3的为主的生成物.在Ⅱc还原过程中,光学混频信号再度减弱,并在这一过程结束时基本回复到整个氧化还原循环初始的信号强度,意味着循环中的氧化产物已被全部还原.最终,Ⅲc对应于界面双电层的放电过程.
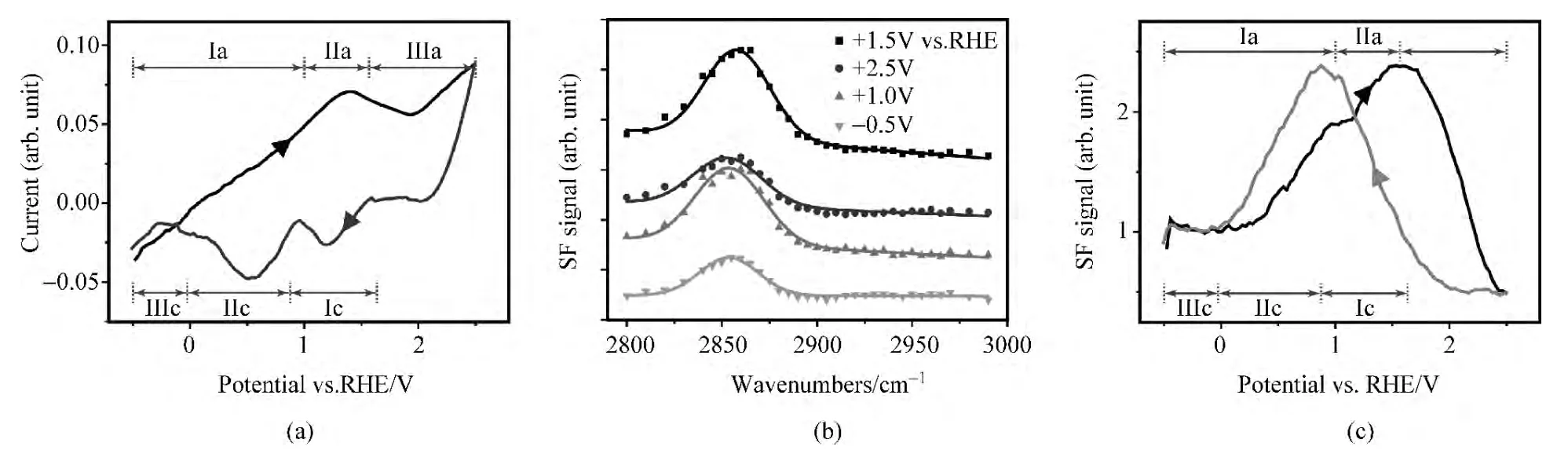
图4 (a)纯金工作电极在pH=12氢氧化钾重水溶液中的循环伏安曲线;(b)该电极的原位和频光谱随偏压变化;(c)2865cm-1混频信号随偏压变化(摘自文献[77])
金表面所吸附的硫醇分子可形成高度有序的自组装膜,而修饰后的硫醇自组装分子层在生物制药、纳米材料领域有大量应用,故而该类体系在基础研究学界也一直备受关注[82].20世纪90年代,Porter等发现了金、铂等贵金属电极表面的硫醇自组装分子层可在随电极表面的偏压变化而脱附与重新吸附.这一发现为硫醇自组装分子层引入了一个新的可控自由度,人们对此开展了大量的研究.然而迄今为止,硫醇自组装分子层在电化学脱附/吸附反应中的具体微观结构仍有诸多争议,对此我们运用表面等离激元增强的光学混频光谱技术进行了研究.图5(a)展示了覆盖有硫醇自组装分子层的金电极在pH=12氢氧化钾重水溶液中的循环伏安曲线,可以清楚地看到在低偏压范围内(0.5~-0.5Vvs.RHE,深色曲线)的硫醇自组装分子层还原脱附峰,相应的化学反应式为:AuSR+e-↔Au+R S-.图5(b)展示了该界面在不同偏压处原位获取的表面等离激元增强的光学混频光谱.在+1.2V,硫醇自组装分子层尚未脱附,其对应的CH3对称伸缩振动模式(~2875cm-1)在光谱上( 曲线)清晰可见,而CH2对称伸缩振动模式(~2850cm-1)则几乎对光谱没有贡献.过去的研究表明,这样的光谱特性标志着由碳氢长链构成的自组装分子层高度有序,结构缺陷几乎可以忽略不计[78].当偏压下降至-0.5V时,循环伏安曲线表明硫醇自组装分子层已全部还原并应从金电极上脱附;然而,来自硫醇分子碳氢键的信号依然十分显著(·表示曲线),意味着脱附后的硫醇分子并未扩散远离电极表面.同时,光谱上的CH3对称伸缩振动模式(~2875cm-1)强度减弱,而CH2对称伸缩振动模式(~2850cm-1)则明显增强,这代表脱附后的硫醇分子层无序度与结构缺陷大量增加.还原脱附后硫醇分子的微观结构对相关过程的各类潜在至关重要,但由于探测手段的限制,对这一问题至今未能有定论[83-87].我们的实时、原位光谱测量表明,还原脱附后的硫醇分子尽管带有负电,却仍聚集在工作电极附近.这应是由于硫醇脱附的同时,溶液中的正离子可以迅速聚集并吸附在工作电极上,处于负偏压的工作电极并未如一些工作所推测的那样将带负电的硫醇分子排斥到远离电极的溶液中.同时,由于实验中所用的长链硫醇分子有较强的链-链相互作用与很低的水溶性,使得脱附后的硫醇分子层仍能保持一定程度上的有序状态.当偏压再度返回到+1.2V处,先期的电化学实验表明硫醇分子可重新吸附回金电极表面并形成自组装层;与此一致的是,该偏压下的光学混频光谱(▲表示曲线)也回复到原来的状态,CH3对称伸缩振动模式(~2875cm-1)再度增强,而CH2对称伸缩振动模式(~2850cm-1)明显减弱,证明重新形成的硫醇自组装分子层与最初制备的自组装分子层同样高度有序,只有很少的结构缺陷.
除了还原脱附外,在较高的正偏压处,硫醇分子也可发生氧化性的脱附,其在强碱性重水溶液中的化学反应式为:
AuSR+2D2O→Au+RS O2D+3e-+3D+
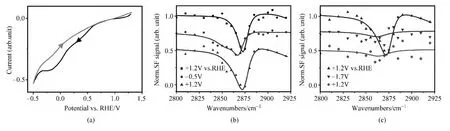
图5 (a)硫醇自组装分子层覆盖的金工作电极在pH=12氢氧化钾重水溶液中的循环伏安曲线;(b),(c)该电极的原位混频光谱在还原与氧化扫描中随偏压的变化(摘自文献[77])
图5(c)展示了该界面在还原曲线不同偏压处原位获取的表面等离激元增强的光学混频光谱.▲表示曲线代表在氧化脱附反应发生之前偏压位于+1.2V的光谱,如上所述.当偏压增加至+1.7V时,上述氧化脱附反应已经发生;与前文所述的还原脱附截然不同都是,同时采集的原位光学混频光谱(▼表示曲线)上几乎没有硫醇分子的振动信号,代表硫醇分子氧化脱附后已离开这一界面.当偏压返回至+1.2V时,同时采集的原位光学混频光谱(◆表示曲线)上仍无硫醇分子的振动信号,表示氧化脱附后的硫醇分子无法重新吸附到工作电极上,主要原因应在于氧化后的硫醇分子可与水分子形成氢键,故有较高的水溶率.
在这一工作中,我们实现了对电化学界面分子振动光谱的实时、原位探测,并研究了两种有着广泛应用的电化学原型体系:纯金电极与水的界面,以及硫醇自组装分子层覆盖的金电极与水的界面.通过光学混频光谱探测,获得了这两种电化学界面的反应中间产物、分子种类与构型等实时信息,为理解这两类电化学反应的微观机制提供了帮助.这一方法可探测各类金属电极与电介质形成的电化学反应,有望在电化学领域获得广泛应用.我们的工作也再次证明,表面等离激元与表面非线性光学的结合确可大大提高实验技术的探测灵敏度,拓宽技术的应用范畴.在未来的研究中,我们计划借助石墨烯等二维材料中的可调谐表面等离激元获取更广频率范围内的场增强效应,以实现更多的光谱学应用.
[1]Shen Y R,Surface nonlinear optics[Invited][J].Journal of the optical society of america B-optical physics,2011,28(2):A56-A66.
[2]Chen C K,Heinz T F,Ricard D,et al.Detection of molecular monolayers by optical second-harmonic generation[J].Physical Review Letters,1981,46(15):1010-1012.
[3]Zhu X D,Suhr H,Shen Y R.Surface vibrational spectroscopy by infrared-visible sum frequency generation[J].Physical Review B,1987,35(8):3047-3050.
[4]Somorjai G A,Rupprechter G.Molecular studies of catalytic reactions on crystal surfaces at high pressures and high temperatures by infrared-visible sum frequency generation(SFG)Surface Vibrational Spectroscopy[J].J.phys.chem.B,1999,103:1623-1630.
[5]Messmer M C,Conboy J C,Richmond G L.Observation of molecular ordering at the liquid-liquid interface by resonant sum frequency generation[J].J.Am.Chem.Soc,1995,117(30):8039-8040.
[6]Ward R N,Davies P B,Bain C D.Orientation of surfac-tants adsorbed on a hydrophobic surface[J].J.Phys.Chem,1993,97(28):7141-7143.
[7]Zhuang X,Miranda P B,Kim D,et al.Mapping molecular orientation and conformation at interfaces by surface nonlinear optics[J].Phys.Rev.B,1999,59(19):12632-12640.
[8]Su X,Cremer P S,Shen Y R,et al.Pressure Dependence(10(-10)-700Torr)of the Vibrational spectra of adsorbed CO on Pt(111)studied by sum frequency generation[J].Phys.Rev.Lett,1996,77(18):3858-3860.
[9]Superfine R,Huang J Y,Shen Y R.Nonlinear optical studies of the pure liquid/vapor interface:Vibrational spectra and polar ordering.[J].Physical Review Letters,1991,66(8):1066-1069.
[10]Du Q,Superfine R,Freysz E,et al.Vibrational spectroscopy of water at the vapor/water interface.[J].Physical Review Letters,1993,70(15):2313-2316.
[11]Du Q,Freysz E,Shen Y R.Surface vibrational spectroscopic studies of hydrogen bonding and hydrophobicity.[J].Science,1994,264:826-828.
[12]Miranda P B,Shen Y R.Liquid Interfaces:A study by sum-frequency vibrational spectroscopy[J].Journal of Physical Chemistry B,1999,103(17):3292-3307.
[13]Ji N,Ostroverkhov V,Tian C S,et al.Characterization of vibrational resonances of water-vapor interfaces by phasesensitive sum-frequency spectroscopy.[J].Physical Review Letters,2008,100(9):1937-1940.
[14]Chen Z,Ward R,Tian Y,et al.Surface composition of biopolymer blends Biospan-SP/Phenoxy and Biospan-F/Phenoxy observed with SFG,XPS,and contact angle goniometry[J].Journal of Physical Chemistry B Materials Surfaces Interfaces Amp Biophysical,1999,103(15):2935-2942.
[15]Zhang D,Gracias D H,Ward R,et al.Surface studies of polymer blends by sum frequency vibrational spectroscopy,atomic force microscopy,and contact angle goniometry[J].J.Phys.Chem.B,1998,102(32):6225-6230.
[16]Zhang D,Shen Y R,Somorjai G A.Studies of surface structures and compositions of polyethylene and polypropylene by IR+visible sum frequency vibrational spectroscopy[J].Chemical Physics Letters,1997,281(4-6):394-400.
[17]Chen C,Liu W,Pagliusi P,et al.Sum-frequency vibrational spectroscopy Study of photoirradiated polymer surfaces[J].Macromolecules,2009,42(6):2122-2126.
[18]Chin R P,Huang J Y,Shen Y R,et al.Interaction of atomic hydrogen with the diamond C(111)surface studied by infrared-visible sum-frequency-generation spectroscopy.[J].Phys Rev B,1995,52(8):5985-5995.
[19]Su X,Cremer P S,Shen Y R,et al.High-pressure CO oxidation on Pt(111)monitored with infrared61Visible sum frequency generation(SFG)[J].Journal of the American Chemical Society,1997,119(17):3994-4000.
[20]Wei X,Miranda P B,Shen YR.Surface vibrational spectroscopic study of surface melting of ice.[J].Physical Review Letters,2001,86(8):1554-1557.
[21]Liu W T,Shen Y R.Surface vibrational modes of alphaquartz(0001)probed by sum-frequency spectroscopy.[J].Physical Review Letters,2008,101(1):4962-4964.
[22]Wang J,Buck S M,Even M A,et al.Molecular responses of proteins at different interfacial environments detected by sum frequency generation vibrational spectroscopy.[J].Journal of the American Chemical Society,2002,124(44):13302-13305.
[23]Liu J,Conboy J C.Direct measurement of the transbilayer movement of phospholipids by sum-frequency vibrational spectroscopy.[J].Journal of the American Chemical Society,2004,126(27):8376-8377.
[24]Liu J,Conboy J C.Phase transition of a single lipid bilayer measured by sum-frequency vibrational spectroscopy.[J].Journal of the American Chemical Society,2004,126(1):8894-8895.
[25]Nguyen K T,Soong R,Lm S C,et al.Probing the spontaneous membrane insertion of a tail-anchored membrane protein by sum frequency generation spectroscopy.[J].Journal of the American Chemical Society,2010,132(43):15112-15115.
[26]Castro A,Sitzmann E V,Zhang D,et al.Rotational relaxation at the air/water interface by time-resolved second harmonic generation[J].J.Phys.Chem,1991,(18):6752-6753.
[27]Guyot-Sionnest P.Coherent processes at surfaces:Free-induction decay and photon echo of the Si-H stretching vibration for H/Si(111).[J].Physical Review Letters,1991,66(11):1489-1492.
[28]McGuire J A,Shen Y R.Ultrafast vibrational dynamics at water interfaces.[J].Science,2006,313(5795):1945-1948.
[29]Smits M,Ghosh A,Sterrer M,et al.Ultrafast vibrational energy transfer between surface and bulk water at the airwater interface.[J].Phys.Rev.Lett,2007,98(9).
[30]Shen Y R.Proceedings of the international school of physics,enrico fermi[Z].Amsterdam:North-Holland,1994.
[31]Shen Y R.The principles of nonlinear optics[Z].New York:Wiley-lnterscience,1984.
[32]Shen Y R,Ostroverkhov V.Sum-frequency vibrational spectroscopy on water interfaces:polar orientation of water molecules at interfaces.[J].Chemical Reviews,2006,106(25):1140-1154.
[33]Wei X,Zhuang X,Hong S C,et al.Sum-frequency vibrational spectroscopic study of a rubbed polymer surface[J].Physical Review Letters,1999,82(21):4256-4259.
[34]Sommerfeld A.The broadening of the waves and the wireless telegraph[J].Annalen Der Physik,1909,28:665-736.
[35]Kretschm E.The determination of the optical constants of metals by excitation of surface plasmons[J].Zeitschrift Fur Physik 1971,241:313.
[36]Otto A.Excitation of nonradiative surface plasma waves in silver by the method of frustrated total reflection[J].Ieitschrift Für Physik A Hadrons & Nuclei,1968,216(4):398-410.
[37]Billmann J,Otto A.Experimental evidence for a local mechanism of surface enhanced Raman scattering[J].Applications of Surface Science,1980,6(80):356-361.
[38]Fleischmann M,Hendra P J,McQuilla A J,Raman-Spectra of pyridine adsorbed at a silver electrode[J].Chemical Physics Letters 1974,26(2):163-166.
[39]Nie S M,Emery S R.Probing single molecules and single nanoparticles by surface-enhanced Raman scattering[J].Science,1997,275(5303):1102-1106.
[40]Zhang J,Fu Y,Chowdhury M H,et al.Metal-enhanced single-molecule fluorescence on silver particle monomer and dimer:Coupling effect between metal particles[J].Nano Letters,2007,7(7):2101-2107.
[41]Brockman J M,Nelson B P,Corn R M.Surface plasmon resonance imaging measurements of ultrathin organic films[J].Annual Review of Physical Chemistry,2000,51:41-63.
[42]Raether H.Surface Plasmons on Smooth and Rough Surfaces and on Gratings[M].Springer-Verlag,1986.
[43]Kauranen M,Zayats A V.Nonlinear plasmonics[J].Nature Photonics,2012,6:737-748.
[44]Chen C K,DeCastro A,Shen Y R,et al.Surface coherent anti-stokes Raman spectroscopy[J].Physical Review Letters,1979,43(13):946-949.
[45]Lamprecht B,Krenn J R,Leitner A,et al.Resonant and off-resonant light-driven plasmons in metal nanoparticles studied by femtosecond-resolution third-harmonic generation[J].Physical Review Letters,1999,83(21):4421-4424.
[46]Lippitz M,van Dijk MA,Orrit M.Third-harmonic generation from single gold nanoparticles.[J].Nano Letters,2005,5(4):799-802.
[47]Palomba S,Novotny L.Nonlinear excitation of surface plasmon polaritons by four-wave mixing.[J].Physical Review Letters,2008,101(5):1603-1606.
[48]Renger J,Quidant R,Hulst N V,et al.Free-space excitation of propagating surface plasmon polaritons by nonlinear four-wave mixing[J].Physical Review Letters,2009,103(26):2725-2727.
[49]Harutyunyan H,Palomba S,Renger J,et al.Nonlinear dark-field microscopy[J].Nano Letters,2010,10(12):5076-5079.
[50]Genevet P,Tetienne J P,Gatzogiannis E,et al.Large en-hancement of nonlinear optical phenomena by plasmonic nanocavity gratings[J].Nano Letters,2010,10(12):4880-4883.
[51]Liao H B,Xiao R F,Fu J S,et al.Origin of third-order optical nonlinearity in Au:SiO(2)composite films on femtosecond and picosecond time scales.[J].Optics Letters,1998,23(5):388-390.
[52]Renger J,Quidant R,Hulst N V,et al.Surface-enhanced nonlinear four-wave mixing.[J].Physical Review Letters,2010,104(4):19-63.
[53]Flytzanis C,Hache F,Klein M.C,et al.Nonlinear optics in composite materials.I:Semiconductor and metal crystallites in dielectrics[J].Progress in Optics,1991,29:321-411.
[54]Danckwerts M,Novotny L.Optical frequency mixing at coupled gold nanoparticles.[J].Physical Review Letters,2007,98(2).
[55]Nathaniel K.Grady,Mark W.Knight,Rizia Bardhan,et al.Optically-driven collapse of a plasmonic nanogap selfmonitored by optical frequency mixing[J].Nano Lett,2010,10(4):1522-1528.
[56]Wang Y,Lin C Y,Nikolaenko A.Four-wave mixing microscopy of nanostructures[J].Advances in Optics &Photonics,2011,3(1):1-52.
[57]Chen C K,Decastro A R B,Shen Y R.Surface-Enhanced Second-Harmonic Generation [J].Physical Review Letters,1981(46):145-148.
[58]Antoine R,Pellarin M,Palpant B,et al.Surface plasmon enhanced second harmonic response from gold clusters embedded in an alumina matrix[J].Journal of Applied Physics,1998,84(8):4532-4536.
[59]Schön P,Bonod N,Devaux E,et al.Enhanced secondharmonic generation from individual metallic nanoapertures.[J].Optics Letters,2010,35(23):4063-4065.
[60]Nieuwstadt J A V,Sandtke M,Harmsen R H,et al.Strong modification of the nonlinear optical response of metallic subwavelength hole arrays[J].Physical Review Letters,2006,97(14).
[61]Chen K,Durak C,Heflin J R,et al.Plasmon-enhanced second-harmonic generation from ionic self-assembled multilayer films.[J].Nano Letters,2007,7(2):254-258.
[62]Fan W,Zhang S,Panoiu N C,et al.Second harmonic generation from a nanopatterned isotropic nonlinear material[J].Nano Letters,2006,6(5):1027-1030.
[63]Cai W,Vasudev A P,Brongersma M L.Electrically controlled nonlinear generation of light with plasmonics.[J].Science,2011,333(6050):1720-.
[64]Ishifuji M,Mitsuishi M,Miyashita T.Bottom-up design of hybrid polymer nanoassemblies elucidates plasmon-enhanced second harmonic generation from nonlinear optical dyes.[J].J.Am.Chem.Soc,2009,131(12):4418-24.
[65]Sanatinia R,Swillo M,Anand S.Surface second-harmonic generation from vertical gap nanopillars[J].Nano Letters,2012,12(2):820-826.
[66]Valev V K.Characterization of nanostructured plasmonic surfaces with second harmonic generation[J].Langmuir,2012,28(44):15454-15471.
[67]Klein MW,Enkrich C,Wegener M,et al.Second-harmonic generation from magnetic metamaterials.[J].Science,2006,313:502-504.
[68]Linden S,Niesler F B P,Forstner J,et al.Collective effects in second-harmonic generation from split-ring-resonator arrays[J].Physical Review Letters,2012,109.
[69]Baldelli S,Eppler A S,Anderson E,et al.Surface enhanced sum frequency generation of carbon monoxide adsorbed on platinum nanoparticle arrays[J].Journal of Chemical Physics,2000,113(13):5432-5438.
[70]Humbert C,Busson B,Abid J P,et al.Self-assembled organic monolayers on gold nanoparticles:A study by sumfrequency generation combined with UV-vis spectroscopy[J].Electrochimica Acta,2005,50(15):3101-3110.
[71]Li Q,Kuo C W,Yang Z,et al.Surface-enhanced IR-visible sum frequency generation vibrational spectroscopy.[J].Physical Chemistry Chemical Physics,2009,11(18):3436-42.
[72]Tourillon G,Laurent Dreesen,Volcke C,et al.Closepacked array of gold nanoparticles and sum frequency generation spectroscopy in total internal reflection:aplatform for studying biomolecules and biosensors[J].Journal of Materials Science,2009,44(24):6805-6810.
[73]Lis D,Caudano Y,Henry M,et al,Selective plasmonic platforms based on nanopillars to enhance vibrational sumfrequency generation spectroscopy.Advanced Optical Materials,2013,1(3):244-255.
[74]Duan J,Park K,Maccuspie R I,et al.Optical properties of rodlike metallic nanostructures:Insight from theory and experiment[J].J.Phys.Chem.C,2009,113(35):15524-15532.
[75]Chen Z,Zhang Z.Enhanced surface sum frequency generation from LB layer covered silver film[J].Journal of Applied Physics,1991,69(11):7406-7410.
[76]Ham E W M,Vrehen Q H E,Eliel E R,et al.Giant enhancement of sum-frequency yield by surface-plasmon excitation[J].Journal of the Optical Society of America B-Optical Physics,1999,16(7):1146-1152.
[77]Liu W T,Shen Y R,In situ sum-frequency vibrational spectroscopy of electrochemical interfaces with surface plasmon resonance[J].Proceedings of the National Academy of Sciences,2014,111(4):1293-1297.
[78]Liu W,Zhang L,Shen Y R.Interfacial layer structure at alcohol/silica interfaces probed by sum-frequency vibrational spectroscopy[J].Chemical Physics Letters,2005,412(1-3):206-209.
[79]Abanulo J C,Harris R D,Sheridan A K,et al.Waveguide surface plasmon resonance studies of surface reactions on gold electrodes[J].Faraday Discussions,2002,121(1):139-152.
[80]Petrovic',M.Metikoš-Hukovic',R.Babic',J.Katic',et al.A multi-technique study of gold oxidation and semiconducting properties of the compactα-oxide layer[J].Journal of Electroanalytical Chemistry 2009,629(1-2):43-49.
[81]Burke L D,Nugent P F.The electrochemistry of gold:II the electrocatalytic behaviour of the metal in aqueous media[J].Gold Bulletin,1998,31(2):39-50.
[82]Love J C,Estroff L A,Kriebel J K,et al.Self-assembled monolayers of thiolates on metals as a form of nanotechnology[J].Chemical Reviews,2005,36(32):1103-1169.
[83]Pesika N S,Stebe K J,Searson P C.Kinetics of desorption of alkanethiolates on gold[J].Langmuir,2006,22(8):3474-3476.
[84]Quinn B M,Kontturi K.Reductive desorption of thiolate from monolayer protected gold clusters.[J].J.Am.Chem.Soc,2004,126(23):7168-7169.
[85]Ron H,Rubinstein I.Self-assembled monolayers on oxidized metals.3.alkylthiol and dialkyl disulfide assembly on gold under electrochemical conditions[J].Journal of the American Chemical Society,1998(120):13444-13452.
[86]Widrig C A,Chung C,Porter M D .The electrochemical desorption of n-alkanethiol monolayers from polycrystalline Au and Ag electrodes[J].Journal of Electro analytical Chemistry,1991(310):335-359.
[87]Yang D F,Wilde C P,Morin M.Studies of the electrochemical removal and efficient reformation of a monolayer of hexadecanethiol self-assembled at an Au(111)single crystal in aqueous solutions[J].Langmuir,1997,13(2):243-249.
