PPARγ介导卡托普利改善高糖诱导血管内皮细胞胰岛素抵抗的作用
2015-06-09严国强陈春香陈芳辉储佳佳黄起壬
严国强,陈春香,陈芳辉,高 艳,储佳佳,李 腾,黄起壬
(1.江西省基础药理学重点实验室,2.南昌大学药学院药理学教研室,江西 南昌 330006)
PPARγ介导卡托普利改善高糖诱导血管内皮细胞胰岛素抵抗的作用
严国强1,2,陈春香1,2,陈芳辉1,2,高 艳1,2,储佳佳1,2,李 腾1,2,黄起壬1,2
(1.江西省基础药理学重点实验室,2.南昌大学药学院药理学教研室,江西 南昌 330006)
目的 研究卡托普利(captopril,Cap)对高糖(high glucose,HG,33 mmol·L-1)诱导的人脐静脉内皮细胞(human umbilical vein endothelium cells,HUVECs)胰岛素抵抗(insulin resistance,IR)的作用及机制。方法 首先观察Cap对HG(33 mmol·L-1)诱导的HUVECs IR的改善作用。实验随机分为5组,即正常对照(Control)组、IR组、IR+CapⅠ(1×10-6mol·L-1)组、IR+CapⅡ(1×10-5mol·L-1)组、IR+CapⅢ(1×10-4mol·L-1)组。其次证实Cap改善HG(33 mmol·L-1)诱导HUVECs IR的作用是由PPARγ介导。实验随机分为6组,即Control组、IR组、IR+Cap(1×10-5mol·L-1)组、PPARγ抑制剂GW9662(PI,1.0 μmol·L-1)组、IR+PPARγ抑制剂(IR+PI,1.0 μmol·L-1)组、IR+Cap+PPARγ抑制剂(IR+Cap+PI,1.0 μmol·L-1)组,除Control组和PI组外,所有各组先用含33 mmol·L-1葡萄糖的DMEM培养48 h,Cap各组再加不同浓度的Cap处理4 h,最后胰岛素(100 nmol·L-1)处理30 min,抑制剂组再加抑制剂(1.0 μmol·L-1)处理1 h,最后进行指标检测。结果 IR组NO水平明显降低、ET-1含量明显升高,提示细胞已产生IR,但PPARγ mRNA和蛋白表达水平与Control组相比差异无统计学意义(P>0.05);Cap呈浓度依赖性逆转HG诱导NO和ET-1水平的改变,明显增加磷酸化PPARγ(P-PPARγ)水平,说明其可明显改善HG诱导的IR,但对PPARγ mRNA和蛋白表达水平无明显影响(vsIR,P>0.05);加PI处理后,Cap改善IR的作用完全取消,提示Cap改善IR的作用是由PPARγ介导。结论 Cap可通过PPARγ介导改善高糖所致血管内皮细胞IR,其机制可能与PPARγ表达水平无关,而与PPARγ激活有关。
卡托普利;高糖;人脐静脉内皮细胞;胰岛素抵抗;PPARγ;NO;ET-1
现代医学研究表明,血管内皮胰岛素抵抗是糖尿病发生血管并发症的始动环节,是滋生多种代谢相关疾病的共同“土壤”[1-2]。但目前,国内外对其发病机制尚不完全清楚。因此,阐明血管内皮IR发生的分子机制,发现糖尿病防治药物新的分子作用靶点及开发新的防治药物,对于降低糖尿病患者心脑血管事件发生,改善患者生存质量具有重要意义。PPARγ 是过氧化物增殖酶体激活受体超家族(PPARs) 的成员之一,是体内糖代谢和脂肪细胞分化的重要调节因子,是胰岛素增敏剂噻唑烷二酮类的靶标[3],能改善血管内皮细胞对胰岛素的敏感性,机制可能与其激活了PPARγ依赖的胰岛素信号转导途径有关。另外,PPARγ也能在配体依赖方式下通过抑制其他转录因子,如NF-κB及活化剂蛋白-1家族,从而直接地抑制促炎症基因的表达[4-5]。研究证实前列腺素衍生物如15-脱氧前列腺素J2(15-d-PGJ2)是PPARγ的天然激动剂[6-7]。
卡托普利(captopril,Cap)是第一代的血管紧张素转化酶抑制剂,临床广泛用于合并了糖尿病的高血压或心衰患者的治疗。有研究证实,Cap可通过增加前列腺素衍生物、缓激肽(bradykinin, BK)水平来改善IR[8-9],那么,它能不能通过直接改变PPARγ的表达或活性来改善IR,目前相关报道甚少,因此,本实验探讨卡托普利改善内皮细胞胰岛素抵抗的机制,为寻找糖尿病血管病变防治的分子靶点拓展新的思路。
1 材料与方法
1.1 细胞株及血清培养基实验用HUVECs株购自美国ATCC(Catalog No: CRL-1730);胎牛血清购自杭州四季青生物工程材料有限公司;DMEM培养基购自Gibco BRL公司。
1.2 药品及试剂NO检测试剂盒购自Beyotime Institute of Biotechnology;ET-1试剂盒购自上海RD生物公司;卡托普利购自上海普康公司;PPARγ抑制剂(GW9662)购自美国Sigma;PPARγ多克隆抗体购自美国Cell Signaling;PPARγ磷酸化抗体购自美国Santa Cruz Biotechnology;β-actin多克隆抗体购自美国Santa Cruz;TRIzol 试剂购自Invitrogen公司;MMLV逆转录酶、RNA酶抑制剂、dNTP、Oligo(dT)15、Taq酶均购自Promega Corporation;引物购自上海捷瑞生物公司;其它各种化学试剂为进口或国产分析纯试剂。
1.3 实验分组及处理方法
1.3.1 Cap对HG诱导的HUVECs IR的改善作用 将HUVECs均匀接种在六孔培养板上,随机分为5组,即Control组、IR组、IR+CapⅠ(1×10-6mol·L-1)组、IR+CapⅡ(1×10-5mol·L-1)组、IR+ CapⅢ(1×10-4mol·L-1)组。除Control组(5.5 mmol·L-1葡萄糖的DMEM培养48 h)外,所有各组先用含33 mmol·L-1葡萄糖的DMEM培养48 h,Cap各组再加不同浓度的Cap处理4 h,最后胰岛素(100 nmol·L-1)处理30 min,收集细胞上清液和细胞,分别进行指标检测。
1.3.2 Cap改善HG诱导HUVECs IR的作用是由PPARγ介导 将HUVECs均匀接种在六孔培养板上,随机分为6组,即Control组、IR组、IR+CapⅡ(1.0×10-5mol·L-1)组、PPARγ抑制剂GW9662(PI,1.0 μmol·L-1)组、IR+PPARγ抑制剂(IR+PI,1.0 μmol·L-1)组、IR+Cap(1.0×10-5mol·L-1)+ PPARγ抑制剂(IR+Cap+PI,1.0 μmol·L-1)组,除Control组和PI组(5.5 mmol·L-1葡萄糖的DMEM培养48 h)外,所有各组先用含33 mmol·L-1葡萄糖的DMEM培养48 h,Cap各组再加不同浓度的Cap处理4 h,最后胰岛素(100 nmol·L-1)处理30 min,抑制剂组再加抑制剂GW9662(1.0 μmol·L-1)处理1 h,收集细胞上清液和细胞,分别进行以下指标检测。
1.4 HUVECs中NO、ET-1水平的检测(a) 参照碧云天生物技术研究所一氧化氮检测试剂盒的使用说明,先做出标准曲线,然后根据各组吸光度,计算出各组NO的含量。(b) 参照人内皮素(ET)酶联免疫试剂盒的使用说明,先做出标准曲线,然后根据各组吸光度,计算出各组ET-1的含量。
1.5 Western blot检测HUVECs内PPARγ、P-PPARγ蛋白的表达细胞经实验因素处理后,经PBS清洗后,用RIPA裂解液进行裂解,蛋白浓度用BCA试剂盒进行测定,细胞裂解物溶于上样缓冲溶液煮沸后经10%的SDS-PAGE电泳分离,并电转到PVDF膜上,取出后将膜放入质量浓度均为50 g·L-1的脱脂牛奶或者BSA中封闭2 h,再用TBST洗膜3次,每次15 min。将膜放入一抗中(1 ∶750),4℃孵育过夜。TBST冲洗膜后,将膜放入相应的二抗(1 ∶2 000)中,室温平摇2 h后,漂洗3次,每次20 min,用ECL化学发光法进行检测。将曝光条带进行X光胶片扫描后,在医学图形分析系统ImageTool软件上进行分析,将所得PPARγ蛋白带的综合密度分别除以对应组β-actin的综合密度后,Excel表中进行分析。
1.6 HUVECs PPARγ mRNA表达检测PCR扩增所用引物由上海生物工程有限公司合成,PPARγ上游引物:5′-TCTGGCCACCAACTTTGGG-3′;下游引物:5′-CTTCACAAGCATGAACTCCA-3′,PCR扩增片段长度为360 bp。内参β-actin上游引物:5′-CGGGAAATCGTGCGTGAC-3′;下游引物:5′-TGGAAGGTGG ACAGCGAGG-3′ ,PCR扩增片段长度为268 bp。PCR扩增体系为25 μL,其中cDNA为5 μL,上、下游引物各1.0 μL,2X Tag酶为12.5 μL,再用无菌三蒸水补至25.0 μL。PCR循环参数为30,94℃预变性5 min,94℃变性45 s,扩增PPARγ为61℃退火30 s,扩增β-actin 为55℃退火45 s,72℃延伸45 s,最后72℃延伸5 min。取扩增产物6 μL上样,1.5%琼脂糖凝胶电泳后,在透射紫外光分析仪下摄影,并在电脑上用Image Tool图像处理软件进行处理。

2 结果
2.1 Cap呈浓度依赖性改善HG诱导HUVECs胰岛素抵抗血管内皮IR评价指标常用血清或培养上清液中NO 和ET-1水平来表示。IR组与Control组比较,NO水平明显降低、ET-1水平明显升高(P<0.01vsControl);用低剂量的卡托普利处理4 h后,与IR相比NO水平明显升高且差异有显著性(P<0.05vsIR),而ET-1水平与IR组比较组间差异没有统计学意义(P>0.05);中、高剂量的卡托普利处理后,NO升高更明显(P<0.01vsIR),ET-1水平明显降低(P<0.05),见Fig 1。
2.2 Cap改善HG诱导HUVECs胰岛素抵抗是由PPARγ介导见Fig 2。PI组与Control组比较,NO、ET-1水平无明显差异(P>0.05);IR+PI组与IR组比较,NO、ET-1水平差异也无统计学意义(P>0.05);IR+Cap组与IR组相比NO水平明显升高、ET-1水平明显下降(P<0.01);而IR+Cap+PI组与IR+Cap组相比NO水平明显下降、ET-1水平明显升高(P<0.01)。

Fig 1 NO,ET-1 levels of various treatments in cultured ±s,n=6)
**P<0.01vscontrol group;#P<0.05,##P<0.01vsIR group

Fig 2 NO,ET-1 levels of various treatments in cultured HUVECs ±s,n=6)
**P<0.01vscontrol group;##P<0.01vsIR group;△△P<0.01vsIR+Cap group
2.3 Cap对HUVECs胰岛素抵抗 时PPARγ mRNA表达的影响PPARγ mRNA各组样本均由对应的β-actin条带的扫描值标化再与Control组比较。PPARγ mRNA在正常HUVECs中呈基础性表达;HUVECs暴露在高糖培养液中48 h后,PPARγ mRNA的表达与Control组比较,差异无统计学意义(P>0.05)。Cap组中内皮细胞的PPARγ mRNA的表达量与IR组相比组间无明显差异(P>0.05),见Fig 3。
2.4 Cap对HUVECs胰岛素抵抗时PPARγ和P-PPARγ蛋白表达的影响PPARγ各组样本均由对应的β-actin条带的扫描值标化。PPARγ蛋白、P-PPARγ蛋白在正常HUVECs中呈基础性表达。IR组中PPARγ蛋白、P-PPARγ蛋白表达量无明显变化,与 Control组相比,差异无统计学意义(P>0.05);Cap组中内皮细胞的PPARγ 蛋白的表达量与IR组相比亦无明显差异(P>0.05);而低、中剂量Cap组中内皮细胞的P-PPARγ 蛋白的表达量较IR明显升高,差异具有统计学意义(P<0.05);而高剂量Cap组与IR组相比P-PPARγ蛋白的表达无明显差异(P>0.05),见Fig 4。
3 讨论
卡托普利可以改善胰岛素抵抗,已有大量的资料证实。可能的作用机制目前有以下几个方面:通过扩血管效应,增加骨骼肌的血流,可促使葡萄糖和胰岛素向胰岛素敏感组织释放, 增加葡萄糖的利用;通过改善胰腺的血液循环从而提高胰岛β细胞的代谢,促进胰岛素分泌,加强靶细胞功能,提高胰岛素的生物效应,改善胰岛素抵抗[10-11];Captopril还能通过抑制血管紧张素转换酶活性,使组织内BK降解减少,局部血管BK浓度增高,BK具有扩张血管和降压作用,BK是血管内皮L-精氨酸-NO途径的重要激活剂,它作用于内皮的β2受体能引起血管内皮超极化因子及NO的释放,BK降解减少还能进一步促进15-d-PGJ2合成,而15-d-PGJ2是PPARγ的天然激动剂。

Fig 3 Effect of various treatments on expression levels of
The left panel shows a representative electrophoretic diagram for PPARγ, β-actin mRNA by agrose gels; the right panel shows a statistical diagram of grey density
在我们的前期工作中,从整体动物、细胞和分子水平观察了PPARγ经典配体如罗格列酮(RG)或匹格列酮(PG)对高糖诱导血管内皮胰岛素抵抗的作用,探讨了其可能的作用机制。结果表明,无论RG或PG预处理和后处理,PG和RG均能明显改善HG诱导HUVEC胰岛素抵抗,其机制是通过非经典PPARγ依赖的NF-κB转录抑制途径(under revision)。PPARγ表达很广泛,包括脂肪细胞、内皮细胞、血管平滑肌细胞、巨噬细胞和心肌细胞等[12]。
本实验中,卡托普利改善胰岛素抵抗是否通过PPARγ来介导的目前机制还并不清楚。实验中用高糖(33 mmol·L-1)处理内皮细胞48 h后再用胰岛素处理0.5 h,取其细胞上清测NO和ET-1水平,发现其NO水平比正常组明显下降,且差异具有统计学意义;ET-1水平明显升高,且差异具有统计学意义。证明胰岛素抵抗的模型建立成功。胰岛素抵抗模型建立后,在预实验中,我们设计了卡托普利浓度梯度(10-8~10-2mol·L-1),结果发现,浓度≤10-7mol·L-1时,Cap对细胞活力和胰岛素抵抗程度均没有影响;而浓度≥10-3mol·L-1时,细胞活力明显下降,表现出明显地细胞毒性。因此,我们就选择了1.0×10-5mol·L-1的浓度。用Cap处理4 h后,其NO水平比IR组明显升高,ET-1水平比IR组明显降低,PPARγ蛋白表达水平与IR组相比无明显改变,但P-PPARγ蛋白表达水平比IR组升高。结合文献资料可能存在机制:在高糖和胰岛素的刺激下,内皮细胞受损,导致NO的分泌减少,ET-1的分泌增多,而这两类物质动态平衡维持着血管的正常状态和功能。用Cap处理4 h后,Cap可以使内皮细胞内BK的减少,进而促进15-d-PGJ2的生成,15-d-PGJ2是PPARγ的天然激动剂,可以使PPARγ磷酸化途径被激活,进而改善胰岛素抵抗。
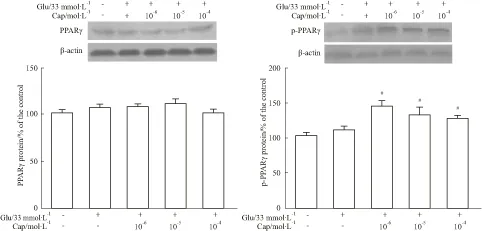
Fig 4 Effect of various treatments on expression levels of PPARγ and P-PPARγ in cultured HUVECs ±s,n=6)
The upper panel shows representative Western blot for PPARγ, P-PPARγ and β-actin; the lower panel shows a statistical diagram of grey density.#P<0.05vsIR group.
综上所述,卡托普利可以促使P-PPARγ蛋白的表达上调,PPARγ与胰岛素信号之间亦存在着正反馈机制;PPARγ可通过转录激活作用增加靶基因的表达或激活来发挥作用。活化的PPARγ能抑制脂肪细胞表达肿瘤坏死因子-α(TNF-α),减轻TNF-α诱发的胰岛素抵抗,且通过增加c-CBL相关蛋白及胰岛素受体底物2的表达,增强胰岛素的信号转导,从而改善胰岛素抵抗。
[1] Ford E S. Risks for all-cause mortality, cardiovascular disease and diabetes associated with the metabolic syndrome: a summary of the evidence [J].DiabetesCare, 2005, 28(7): 1769-78.
[2] Marina C, Maria A M, Simona F,et al. Carotid artery intima-media thickness is associated with Insulin-mediated glucose disposal in nondiabetic normotensive offspring of type 2 diabetic patients [J].AmJPhysiolEndocrinolMetab, 2007, 292(1): E347-52.
[3] 张 宁,孟爱民,王莉莉.选择性PPARγ调节剂治疗二型糖尿病的分子机制研究进展[J].中国药理学通报,2013, 29(2):157-60.
[3] Zhang N, Meng A M, Wang L L. Molecular mechanism of selective PPARγ modulators for type 2 diabetes treatment[J].ChinPharmacolBull, 2013, 29(2):157-60.
[4] Veliceasa D, Schulze-Hoepfne Ft, Volpertl O V. PPARγ and agonists against cancer: rational design of complementation treatments [J].PPARRes,2008, 2008:945275.
[5] Patel N G, Holder J C, Smith S A. Differential regulation of lipogenesis and leptin production by independent signaling pathways and rosiglitazone during human adipocyte differentiation [J].Diabetes, 2003, 52(1):43-50.
[6] Scheen A J. Renin-angiotensin system inhibition prevents type 2 diabetes mellitus part1, a meta-analysis of randomized clinical trials [J].DiabetesMetab, 2004, 30(6): 487-96.
[7] Haseqawa H, Takano H, Komurol. Therapeutic implications of PPARgamma in cardiovascular diseases [J].PPARRes, 2010, 2010:876049.
[8] Sharma J N, Kesavarao U. The effects of captopril on cardiac regression, blood pressure and bradykinin components in diabetic Wistar Kyoto rats[J].ImmunopatholPharmacol, 2011, 24(2): 337-43.
[9] Adam A, Leclair P, Montpas N, Koumbadinga G A. Altered cardiac bradykinin metabolism in experimental diabetes caused by the variations of angiotensin-converting enzyme and other peptidase [J].Neurpeptides, 2010, 44(2): 69-75.
[10] Hermann T S, Li W, Dominguez H, et al. Quinapril treatment increases insulin-stimulated endothelial function and adiponectin gene expression in patients with type 2 diabetes [J].ClinEndocrinolMetab, 2006, 91(3): 1001-8.
[11] Leung P S, Carlsson P O. Tissue renin-angiotensin system:its expression,localization, regulation and potential role in the pancreas [J].MolEndocrinol, 2001, 26(3): 155-64.
[12] Videla L A, Pettinelli P. Misregulation of PPAR functioning and its pathogenic consequences associated with nonalcoholic fatty liver disease in human obesity [J].PPARRes, 2012, 2012:107434.
Improvement effect of captopril on insulin resistance mediated by PPARγ in vascular endothelial cells
YAN Guo-qiang1,2, CHUN Chun-xiang1,2, CHEN Fang-hui1,2,GAO Yan1,2, CHU Jia-jia1,2, LI Teng1,2, HUANG Qi-ren1,2
(1.TheKeyLaboratoryofBasicPharmacologyinJiangxiProvince; 2.DeptofPharmacology,CollegeofPharmacy,NanchangUniversity,Nanchang330006,China)
Aim To investigate the role of captopril in insulin resistance of endothelial cells induced by high glucose. Methods 1. Improvement effect of captopril on insulin resistance in HUVECs was observed. The HUVECs were seeded in a 6-well plate and were randomly divided into 5 groups, namely, control group, IR group, IR together with different Cap concentrations (low, medium and high concentration), respectively. 2. Improvement effect of Cap on insulin resistance was mediated by PPARγ in HUVECs. HUVECs were randomly divided into 6 groups, namely, control group, control+PPARγ inhibitor (PI)(1.0 μmol·L-1)group, IR group, IR+PI(1.0 μmol·L-1)group, IR+Cap( 1×10-5mol·L-1) group, and IR+Cap+PI (1.0 μmol·L-1)group. All indicators were detected. Results After HUVECs were incubated with media containing 33 mmol·L-1of glucose for 48 h, the NO levels were significantly decreased while ET-1 levels were significantly elevated, showing a significant difference between IR group and control group (P<0.01). The expression levels of PPARγ mRNA and its protein were somewhat up-regulated, but there was no significant difference between IR group and control group (P>0.05). When the HUVECs in IR group were treated with DMEM containing glucose (33 mmol·L-1) for 48 h and insulin for 30 min, the expression levels of PPARγ mRNA and its protein in Cap groups were similar to those in the IR group, and there was no significant difference between the two groups (P>0.05); however, the expression levels of phosphorylated-PPARγ protein in Cap groups were increased compared with IR group (P<0.05). The levels of NO were significantly increased whereas the levels of ET-1 were decreased in Cap groups, which had significant differences compared with IR group (P<0.05). Nonetheless, pre-treating with GW9662, a PPARγ inhibitor, the improvement effects of Cap were markedly abolished. Conclusions Captopril could improve high glucose-induced insulin resistance of endothelial cells mediated by PPARγ, and the underlying mechanisms are related to the activation of PPARγ, rather than its expression.
captopril; high glucose; human umbilical vein endothelium cells; insulin resistance; PPARγ; NO; ET-1
时间:2015-3-16 15:41 网络出版地址:http://www.cnki.net/kcms/detail/34.1086.R.20150316.1541.027.html
2014-10-31,
2014-12-04
国家自然科学基金资助项目(No 81360060、81070633、30860111、30660058);江西省主要学科学术和技术带头人培养计划项目(No 20123BCB22005)
严国强(1989-),男,硕士生,研究方向:内分泌代谢性疾病药理学,E-mail:ygq19890926@126.com 黄起壬(1967-),男,博士,教授,博士生导师,研究方向:内分泌代谢性疾病药理学,通讯作者, E-mail:qrhuang@ncu.edu.cn
10.3969/j.issn.1001-1978.2015.04.019
A
1001-1978(2015)04-0532-06
R322.12;R458.5;R543.02;R587.1;R972.4;R977.6
