美国《孤儿药法案》的变迁及启示
——基于对中国生物医药产业的研究
2015-05-20高山行
高山行,韩 晨
(西安交通大学管理学院,陕西西安 710049)
美国《孤儿药法案》的变迁及启示
——基于对中国生物医药产业的研究
高山行,韩 晨
(西安交通大学管理学院,陕西西安 710049)
运用制度理论和现象分析法,探讨了美国《孤儿药法案》形成以及颁布30余年的变迁,重点分析了其立法调整过程以及制度环境对生物医药产业发展的影响;认为其形成和变迁是一个三阶段的社会化过程,与产业组织结构相适应的制度环境能够有效促进产业发展;患者群体、联邦政府和制药企业分别在三个阶段中扮演主导角色,文化-认知性要素、规制性要素和规范性要素分别在三个阶段的变迁中起到推动作用;在结合中国实际的基础上,提出有效促进中国生物医药产业发展的核心是,既要紧抓制度环境建设,完善规制性要素,发挥其对产业发展的法律支柱作用,还要强化文化-认知性要素,夯实其积累基础并提前防范,关注规范性要素,以调控产业良性发展。
美国;孤儿药法案;制度理论;现象分析法;中国生物医药产业
近年来,我国罕见病患者得到了越来越多的关注(如2014年流行的“冰桶挑战赛”对于肌萎缩侧索硬化症①肌萎缩侧索硬化症发病率为十万分之二到十万分之五,和癌症、艾滋病、白血病、类风湿一起被WTO列为全球五大绝症。患者的关注)。2013年,美国《孤儿药法案》(Orphan Drug Act,以下简称《法案》)颁布和国家罕见病组织(National Organization for Rare Disorders,NORD)成立30周年。这30年来,《法案》取得了超乎预想的成就,带来了四个方面的变化:一是孤儿药的注册申请数量大幅度提高,累计3 279种药品获得孤儿药资格认定,489种孤儿药成功上市[1];二是孤儿药成为“重磅炸弹”的潜力充分显现;三是多国政府纷纷效仿美国以立法形式对孤儿药加以促进,如欧盟、新加坡、日本、澳大利亚等;四是跨国公司加大了对孤儿药的研发投入[2]。
本文通过对《法案》形成及演化的研究,探索在无法通过市场机制自行运转、需要政府政策干预和制度设计来推动其运行的产业中,制度的形成和变迁过程,以期对我国生物医药产业的发展提供参考。
一、制度理论概述
制度理论强调制度对于组织决策和组织行为的影响。斯科特将制度定义为:为社会生活提供稳定性和意义的规制性、规范性和文化-认知性要素,以及相关的活动和资源,由符号性要素、社会活动和物质资源构成的持久性的社会结构[3]。规制性要素、规范性要素和文化-认知性要素构成了制度理论的三大支柱,它们以相互独立或相互强化的方式,构成一个强有力的社会框架[3]。
三大支柱在制度变迁过程中所产生的影响是不同的。在不同的阶段,三种要素的地位和作用是差异化的,且会发生变化[4]。制度理论存在于世界系统、社会、组织场域、组织种群、组织以及组织亚系统六大层面。不同层次的制度是相互影响,相互制约的。上一层次的制度提供了下一层次制度的发展背景,而后者也对上一层次的制度产生作用,塑造其生存背景[5]。因此,研究制度变迁有必要将研究构建在多个不同的层次上,对三大支柱和六大层次进行交叉研究。
美国具有清晰的制度体系和价值观系统,而《法案》的变迁过程也是国家立法的缩影。因此,制度理论尤其适用于研究《法案》的变迁。本文研究的是《法案》在与制度环境相互作用中的变迁历程以及制度理论视角下该变迁产生的原因。从范式选择的两条轴线来看,一方面,《法案》的变迁是被其内在秩序和规则所主导的,而不是杂乱无章、随意剧烈的;另一方面,我们将《法案》变迁过程中各行为主体与三种关键制度因素相联系,积极主动地从《法案》这个规则和社会实体的产生、保持和演变的讨论中,提炼出有关内部秩序变迁的主观思考。此外,《法案》变迁是一个复杂和具有多重面相的社会过程,与历史和社会背景密切相关,且随时间发生动态演进。因此,诠释型范式最适用于本文的研究。
诠释型范式中的现象分析法由伯格和卢克曼(Berger and Luckmann)[6]提出,强调主观经验是理解客观现实的方式,通过对与主观经验相关的现象进行系统回顾和分析,以达到归纳出事物演变背后机制和原因的目的[7]。现象研究关注驱动因素、重大事件以及结果三部分,不受事物具体细节的拘束,因此特别适合面向复杂系统的研究。本文着眼于制度环境的发展和变化,以呈现、理解和归纳出《法案》的形成和变迁过程,因此适合采用现象分析法。我们首先对《法案》的内容和影响做简要介绍;接下来将《法案》的演变划分为三个阶段,针对每一阶段,采用现象分析法,归纳其变迁的驱动因素、重大事件以及结果;随后应用制度理论,从联邦政府、制药企业和罕见病患者三级层面上,将变迁各阶段的特征与制度理论的三种基础要素相关联,探讨制度要素的演变及其作用;最后总结研究结果,并据此提出对于我国生物制药产业发展的启示。
本文用于分析的资料来源于:(1)1983年至2014年,美国食品和药物管理局(Food and Drug Administration,FDA)出台的针对孤儿药的相关政策和文件;(2)医疗健康调研公司(EvaluatePharma)发布的2013和2014年孤儿药报告;(3)邓克尔(Dunkle)所著的《孤儿药法案》30年回顾[8];(4)全球法律信息数据库(Lexis.com)中包含的有关美国孤儿药的法规、案例、论文和著作等。多种资料来源相互补充相互验证,以确保资料的准确性和可靠性。
二、美国《孤儿药法案》的变迁历程
孤儿药(orphan drugs)是指用于预防、治疗、诊断罕见病的药物、疫苗、诊断试剂、医疗器械等[9]。目前已发现的罕见病多达6 000到7 000种,占人类疾病总数的10%[10]。孤儿药问题最早起源于美国。20世纪70年代,由于孤儿药市场需求小、研发成本高,制药企业往往难以通过市场机制收回成本,导致制药企业研发积极性低,即使有新的药物研制出来,也不愿意将其投放市场。为了鼓励企业投资研发和生产,1983年1月4日,美国总统里根签署《法案》,开创了孤儿药由政府支持的先河,成为美国罕见病用药方面的基础性法律。
《法案》规定,药物目标患者总数小于20万人的药物为孤儿药[11]。美国政府采取多种法律手段激励企业研发:基金资助;协议帮助;课税扣除;7年市场垄断权;开放性协议;快速审批通道等。这些政策激励收获颇丰。美国食品和药物管理局数据库中孤儿药身份认定和上市的数据显示:截止目前,累计489种孤儿药品种获得批准;而在1983年《法案》颁布以前,仅有38种孤儿药通过审批。图1显示1983年到2014年获得身份认定和成功上市的孤儿药数量①数据来源:美国食品和药物管理局孤儿药审批数据库。http://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm.[1],图2显示2000年至2020年孤儿药销售额以及孤儿药占处方药销售额的比例(2014年后为预测值)②数据来源:医疗健康调研公司(EvaluatePharma)2014年孤儿药研究报告。http://info.evaluategroup.com/rs/evaluatepharmaltd/images/2014OD.pdf.[12]。
根据联邦政府、制药企业以及罕见病患者群体在孤儿药立法的形成和发展中所扮演的角色不同,我们将《法案》变迁的历程分为三个阶段,其中一些里程碑事件归纳见表1。
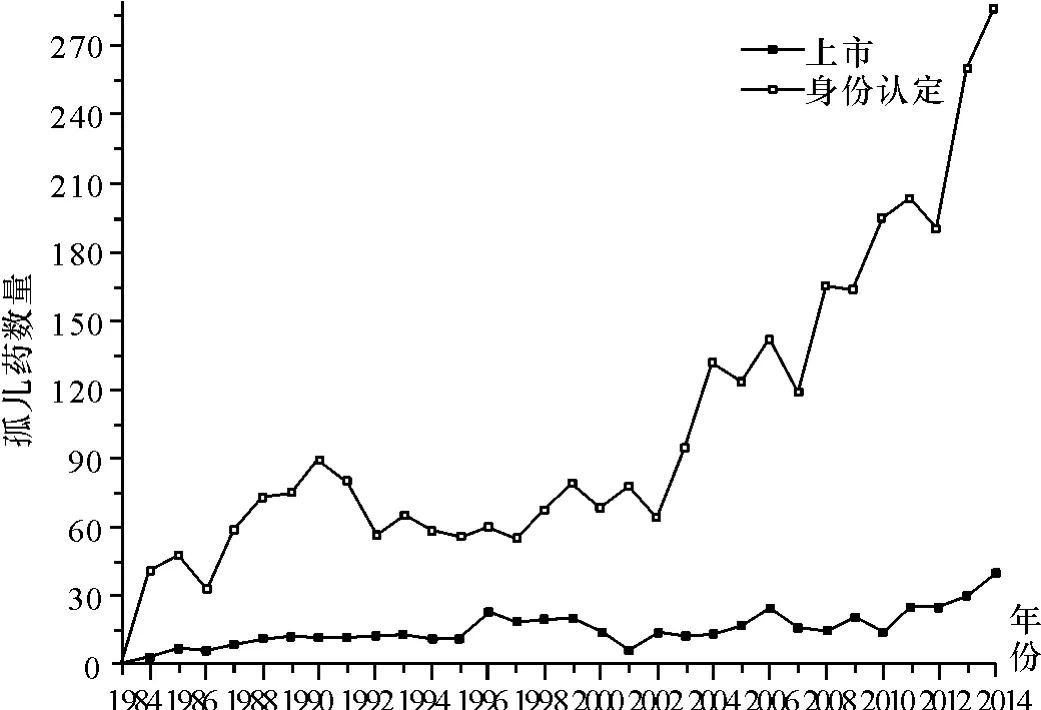
图1 1983-2014年美国孤儿药的上市和身份认定情况
(一)第一阶段:1983年以前
在这一阶段,通过立法手段推动孤儿药研发以解决孤儿药短缺问题的思想处于萌芽阶段,患者群体以各种方式推进将孤儿药纳入立法范畴的进程。
《法案》的建立是一个自下而上的过程,政府公职人员、民众、媒体都参与其中。早在20世纪70年代,美国国立卫生研究院和美国食品和药物管理局(FDA)就已经意识到鼓励孤儿药研发需要系统化有组织的协同行动。但真正驱动孤儿药法案产生的是两起孤儿药短缺事件。
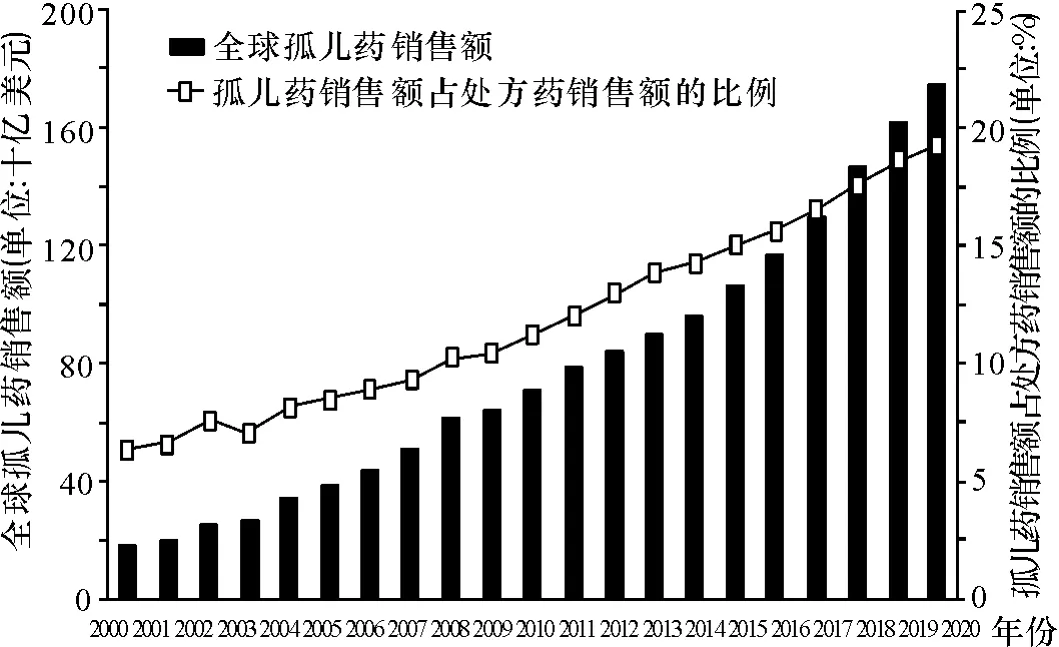
图2 2000-2020年全球孤儿药销售额及其占处方药销售额的比例

表1 美国孤儿药法律体系形成史
1.驱动因素。1979年6月,美国食品和药物管理局(FDA)布报告呼吁政府、企业和公众关注目标患者数量极少的药品的研发和生产问题。与此同时,图雷特综合征和亨丁顿舞蹈症患者因药品短缺,向美国当时的贸易与能源委员会健康与环境分会主席韦克斯曼(Waxman)和堪萨斯州的参议员卡斯鲍姆(Kassebaum)寻求帮助。这两起药品短缺事件触发了《法案》的建立,使政府决定正视孤儿药市场状况。
2.重大事件。1980年6月,韦克斯曼(Waxman)举行面向罕见病群体的听证会,随后对制药企业、联邦政府及其研究机构、食品和药物管理局(FDA)、科研院所等进行大规模市场调查。结果显示,当时共有134个孤儿药品种,其中47个品种被联邦政府批准,而仅有10个品种得以上市;非专利孤儿药无法获利;孤儿药的临床试验难度巨大[13]。韦克斯曼(Waxman)于1981年起草了促进孤儿药研发的草案(立法草案编号:H.R.5238),热播节目“昆西(Quincy ME)”中罕见病患者的演出对该《法案》的通过起到很大的推动作用。国家罕见疾病组织等社会组织对此进行了广泛宣传,最终《法案》于1983年1月4日通过。此外,为了促进罕见病的基础研究,食品和药物管理局(FDA)于1982年设立孤儿药研发办公室,统筹安排全国范围的孤儿药研发认定工作。
3.结果。孤儿药研发办公室的设立以及《法案》的正式出台,标志着全美范围内孤儿药和罕见病立法制度的建立。但是当时《法案》的内容极其简单:只针对少数疾病(例如亨廷顿综合征、鲁·贾里格综合征等),并不能完全涵盖孤儿药和罕见病的范畴;仅包括两条激励条款(临床试验可获得联邦资助,自孤儿药资格获批之日起企业即拥有7年市场专营权)。
(二)第二阶段:1983年至1989年
联邦政府于1984年以后对《法案》进行多次修订。1990年以前,仅仅是针对《法案》疏漏之处的修订,目的是使《法案》更加完善,以更好地激励企业的积极性;而1990年以后,《法案》可能被滥用的风险逐步得到认可。期间举行多次听证会,试图解决企业对于孤儿药定价过高的问题,虽然未曾通过,但却给《法案》的修订敲响了警钟。因此,以1990年第一次探讨高价孤儿药问题的听证会为界,1984至1989年为法案修订的第二阶段,1990年后为第三阶段。
1.驱动因素。第二阶段是法案修订的关键阶段,法案出台后所暴露出来的两大问题促使联邦政府对法案不断修订和完善。第一,《法案》对于孤儿药和罕见病的认定标准不够完善。1983年法案对孤儿药的认定标准是经济指标。制药企业只要提供证据证明某种药品的收益远小于其研发成本,通过美国食品和药物管理局审核后,无论其目标人群规模如何,企业都可获得资金资助和7年市场专营权。这一经济标准对食品和药物管理局和制药企业的药品评估和统计构成了挑战,也造成了不必要的人员浪费。第二,孤儿药7年市场专营权与药品专利期不相容。1983年《法案》仅对《法案》生效前无法取得专利的孤儿药提供7年市场专营权,但是对于部分已经申请了专利的孤儿药,由于研发周期长,其专利在上市后不久甚至上市前就会失效,造成企业收益仍不能抵消其研发开支。
2.重大事件。1984年初,赫氏(Hatch)建议对罕见病设定人群规模限制。同年3月在华盛顿举行关于此修正案的听证会,美国食品和药物管理局与国家罕见疾病组织进行了激励讨论;随后10月,《法案1984》(编号:公共法98~551)正式出台,将罕见病的规模定为不足20万。1985年5月孤儿药法案修订案(H.R.2290)对于全部孤儿药提供7年的市场专营权,无论专利状态。1988年,孤儿药必须先由企业按标准申请并获得孤儿药身份认定,才可以获批上市,并且在原有药品范围外,纳入了生物制品、医疗器械和医用食品,扩大了孤儿药的范畴。
3.结果。经过多次修订,《法案》基本得到固定,其内容和条款能够保证制药企业研发和生产孤儿药有利可图,极大地促进了企业的研发积极性,确保了罕见病患者有药可医。
(三)第三阶段:1990年以后
在继续完善立法条款的同时,联邦政府和患者对《法案》可能被滥用的风险得到初步理性认识,试图对《法案》中可能被滥用的条款进行修订,从而限制制药企业因处于垄断地位、对孤儿药定价过高而获得超额收益。
1.驱动因素。第一,《法案》给予制药企业7年市场专营权,却没有对孤儿药定价做出限制,造成了垄断。例如有些销量不大的药物却因为市场独占,企业可以把其价格定得很高,企业的实际收益远远大于其研发投入。比如生长激素,每个病人的花销在1到3万美元之间,年销售额高接近2亿美元,而实际研发费用只有两三千万。第二,有的药物对多种罕见病起作用,实际销量并不小,但依旧被认定为孤儿药,享受高额研发补贴和市场专营权,企业实际上还是获得了超额收益。第三,还有一些疾病初期满足罕见病的人群规模,后来随着人数越来越多,其实早已不再是罕见病(最典型的就是艾滋病),但是《法案》却没有据此做出调整,依然认定该药物为孤儿药,这远远与《法案》设立的初衷相悖。
2.重大事件。1990至1994年在美国举行多次听证会,探讨如何评估和控制高价孤儿药问题。联邦政府、制药企业、罕见病患者三方均出席并参与。
1990年2月的听证会着重讨论:(1)法案是否造成了不必要的垄断;(2)艾滋病患者数量已经超限,抗艾药是否应该被排除出孤儿药的范围。韦克斯曼(Waxman)认为,防止垄断的措施之一是让生产相似孤儿药的企业共享7年市场专营权。但此提议被乔治·布什总统以监管和实施太困难、企业既得利益受到侵犯、企业积极性受到打击为由拒绝。1992年3月的两次听证会中,多名议员认为法案已经妨碍了良性市场竞争,建议收回累积销售额超过2亿美元的药品的市场专营权。1994年6月的听证会进一步探讨了该问题但却一直未通过。
然而,持续完善《法案》的相关提议均得到了通过。1991年《法案》增加了新申请的药物与已经上市的适用于同一罕见病的药品相比,除非其被证明更加有效,美国食品和药物管理局才会认定其孤儿药资格,消除了企业对搭便车者的防备,促进了药品创新①这一修订的起源是基因泰克(Genentech)和礼来(Eli Lilly)关于儿童生长激素补充剂的争执。基因泰克(Genentech)首先研发出治疗儿童生长激素缺乏的由191个氨基酸序列构成的大分子结构,获得了美国食品和药物管理局(FDA)的孤儿药资格认定,并享有7年市场专营权。随后礼来(Eli Lilly)也研发出了治疗同样疾病的药物,所不同的是其分子结构包含192个氨基酸序列,同样获得了孤儿药资格认定和市场专营权。基因泰克(Genentech)认为两种分子结构并无大的区别,其实多增加的那个氨基酸对于疾病的治疗并无效果,礼来(Eli Lilly)存在搭便车之嫌。基因泰克(Genentech)多次就此提起诉讼,但均以失败告终,因为美国食品和药物管理局(FDA)并无明确关于“相同或相似的分子结构”的定义。。1997年的《减轻税负法案》规定孤儿药临床试验可减免50%税额。2002年出台《罕见病法案》及《孤儿药研究资助法案》;2010年举办了第一届孤儿药审查员培训课程;2013年颁布《孤儿药实施细则》。这一系列的修正案标志着《法案》正在走向成熟。
3.结果。《法案》的变迁仍在继续,各利益相关者对《法案》的评价也愈发理性。总的来讲,《法案》是成功的,具有多方面的积极作用。首先,丰富了孤儿药品种,增加了孤儿药数量,从1983年的38种增至目前的近500种。其次,《法案》赋予孤儿药研发更多便利条件,联邦政府通过多种方式帮助企业进行孤儿药的研发和生产。最后,《法案》促进了制药企业对生物药的研发积极性,使生物医药产业成长为朝阳产业,有助于推动生物医药产业的发展。但是《法案》造成的部分负面影响(如孤儿药高价问题和妨碍市场竞争造成垄断的问题),却一直未得到解决。
三、对美国《孤儿药法案》变迁的讨论
在复杂多变的环境中,将《法案》颁布30余年来的变迁置于历史背景中,关注其在制度理论三大基础要素方面的变化,以及这种变化对于《法案》的作用和影响,我们能够更加清晰地认识和理解美国孤儿药立法的演变。
在《法案》变迁的第一阶段,大多数事件发生在患者群体层面上,主要对固有价值体系产生冲击。患者组织的多次呼吁使政府和社会公众认识到自由竞争的价值观念并不适用于孤儿药,市场调节供给和需求的能力无法满足患者对于孤儿药的需求。在这一阶段,虽然孤儿药议题的共享价值观以及认知框架得到了认可,却没有清晰的法律框架和制度安排去实际执行这种价值体系。价值观真正固化和内嵌于建设性的实践活动,需要明确和完备的规则和法规的指导。虽然孤儿药需要政府扶持和激励的观念早在20世纪70年代以前就已经萌生,但直到1983年才真正以立法的形式规范下来。在新旧价值观和制度安排的冲突之间,出现了一系列矛盾。例如,制药企业对于《法案》的反应远不如政府预想的那么积极。《法案》颁布一年内,只有15起孤儿药资格认定的申请,其中只有10起得到了批准。价值观的扭转也是异常困难的。为了将孤儿药纳入立法保障的范畴,患者群体和政府可谓费尽艰辛。多次游说、媒体宣传和听证会以后,孤儿药的研发和生产终于获得了法律保障。
第一阶段中,变化主要由文化-认知要素所主导,规则和法条并没有得到具体的实践。与此不同的是,第二阶段的制度变迁主要和准则和法规相关。联邦政府首次以立法的形式对如何促进孤儿药研发和生产予以规定,促进孤儿药研发和生产的制度框架首次得到确定,随后又多次针对1983年《法案》中所存在的问题进行了修订。这一阶段,联邦政府颁布了一系列重要的修正案,产业结构发生了变化,原来的供给小于需求正在逐步转变为供给和需求相平衡,促进了药品创新,企业在孤儿药研发中的主动性和投入日益突出。
《法案》发展到第三阶段,主要的法律和法规得到固定,企业在立法框架下的活动和对于立法的反应成为推动《法案》变迁的主要力量。制药企业认识到孤儿药市场的潜在收益,逐步成为更加独立、主动和自主的经济体,但也出现了“搭便车”和“钻空子”的问题。联邦政府考虑到《法案》被滥用的风险,着力于提供更加公平和有利于竞争的制度框架。从第二阶段过渡到第三阶段,制度环境从既不稳定也不均衡变得趋向于稳定和均衡,规范性要素开始在其中扮演重要角色。三个阶段的制度变迁参见表2。

表2 《孤儿药法案》制度变迁表
不同阶段中处于支配地位的角色是不同的,关键的制度要素也是不同的。患者群体、联邦政府和制药企业分别在三个阶段中扮演主导角色;文化-认知性要素、规制性要素和规范性要素分别在三个阶段的变迁中起推动作用见表3。

表3 制度变迁中的支配性角色与关键制度因素
这种主导角色和主导要素的变化说明,制度变迁需要具有不同利益逻辑的多种利益相关者和多种制度要素的共同作用。联邦政府的利益出发点是整个社会的福利与均衡;制药企业是理性的经济体,是利益导向的;而患者群体的福利则是保证药物治疗可及性。可见,联邦政府是均衡企业经济利益和患者福利的一座桥,发挥着协调作用。
四、美国《孤儿药法案》对我国的启示
近年来,我国生物医药产业越来越受到关注。2010年《国务院关于加快培育和发展战略性新兴产业的决定》,2011年《“十二五”生物技术发展规划》,2012年《医药工业“十二五”规划》,2013年《生物产业发展规划》,以及2015年《中国制造2025》等规定均将生物医药产业作为发展的重中之重。然而,目前我国生物医药产业创新不足,企业在自主研发方面心有余而力不足。促进我国生物医药产业的发展,我们认为可以从美国孤儿药立法中得到线索和指导。首先,研发孤儿药和生物技术药物的收益均小于其研发成本,都需要政府干预与引导。其次,从孤儿药自身特点来讲,其患病人群数量小,80%由遗传缺陷引起,且多有家族性倾向,容易导致致病基因的扩散,这就使生物技术药物的研发有了用武之地。再次,从技术手段来讲,孤儿药研发主要依赖生物技术手段。健赞公司(Genzyme Corporation)①全球较早成立的前十大生物制药企业,其产品大都采用生物技术手段来寻求孤儿病的解决方案,被称为“孤儿药之王”,其孤儿药品种多达20余种。所研发的孤儿药大都通过生物技术手段实现。如用于治疗戈谢病的思而赞(注射用伊米苷酶,Cerezyme)是采用基因工程手段利用仓鼠细胞研制而成[14,15]。
产业发展路径由文化-认知性要素主导阶段、规制性要素主导阶段以及规范性要素主导阶段三部分依次衔接而成,与产业组织结构相适应的制度环境能够有效促进产业的发展。
自《国家中长期科学和技术发展规划纲要(2006年-2020年)》颁布以来,我国政府以及非盈利组织对于生物医药产业给予诸多关注,现行文化-认知要素已有一定基础,关注生命与健康的共享价值观得以普及,但却没有配套的法律法规去支撑产业发展。因此,目前,我国发展生物医药产业首要是制度建设与立法支撑。政府应该通过生效最快的规制性要素,将支撑生物制药产业发展的保障措施纳入法律框架,通过立法和政策去引导产业发展,为产业发展提供合法性保障。
在紧抓立法建设的同时,我国仍需强化文化-认知要素。文化-认知要素的发展不是一蹴而就的,而是一个循序渐进、不断持续的过程。目前文化-认知要素的积累已有一定基础但还存在不足,罕见病和孤儿药问题一直未得到应有的关注,直至2014年冰桶试验才被提上日程。此外,发展生物医药产业必须具有前瞻性。《法案》经历30年的变迁依旧存在高价孤儿药、垄断、创新药和仿制药的激励与平衡等悬而未决的难题。因此,我国生物医药产业的发展应提前防范,早做预警。
任何制度的发展和演变都是一个不断调整的周期性过程。从价值观的宣扬,到法律法规的出现,最后必然需要标准和准则去规范制度和产业的发展。因此,当生物制药产业在立法的保障下发展到一定阶段时,可以通过强化和控制规范性要素(如通过企业和行业层面的培训来规范管理和技术准则),调控产业朝着经济和社会需要的方向良性发展。在规制性立法方面,2009年1月9日,国家食品药品监督管理总局(CFDA)颁布实施《新药注册特殊审批管理规定》,提出创新型药物在四种情形下可实行特殊审批②(1)未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂,新发现的药材及其制剂;(2)未在国内外获准上市的化学原料药及其制剂、生物制品;(3)治疗艾滋病、恶性肿瘤、罕见病等疾病且具有明显临床治疗优势的新药;(4)治疗尚无有效治疗手段的疾病的新药。,在激励生物医药产业发展方面迈出了一大步。但与美国促进孤儿药研发的《法案》相比,我国在生物医药产业立法方面,仍有完善的空间,具体可采取以下措施:
(一)鼓励研发生产有针对性生物技术药物的企业
对生物技术药物的临床试验给予政府研究补助金,用于资助I期和II期临床试验;帮助企业进行临床前研究和临床研究,使研究既符合国家食品药品监督管理总局的要求,又能缩短研究时间,降低开发成本;对生物技术药物的研发减免税额,具体的减免额度根据生物技术药物的需求和供给情况进行动态调整;在新药获得专利权之前给予生物技术制药企业一段时期的市场专营权(除非该药供应不足或有比该药更安全、更有效或具有临床优越性的药物出现),不再批准治疗相同疾病的药物上市。
(二)优先考虑将治疗重大疾病的生物技术药物纳入省级和国家医保药品目录
目前,我国医保对生物技术药物的覆盖面过小,患者对于高昂的生物技术药品望而却步。若能够完善生物技术药物进入医保的通道,则患者对生物技术药品的需求能够得到满足,也提高了企业研发生物技术药品的动力。创建国家医保药品目录新类别,以涵盖生物技术药物及其他创新治疗药物。考虑到地区间的差异和此政策执行的复杂度,可先鼓励经济发展水平较高且积极发展生物技术产业的省市率先将生物技术药品纳入本地区的医保目录,再通过长期努力和多方协作,使其逐步扩散到全国,最终形成统一的医保药品目录。
(三)鼓励引进安全、可靠、先进的生物技术药物研发中心和生产基地
我国可通过法律和政策引导,充分利用技术外溢,促进国内生物技术药物研发和生产。
通过法律和政策引导,鼓励将安全、可靠、先进的生物技术药物的研发中心、生产基地引进我国,充分利用其技术外溢,促进国内生物技术药物的研发和生产。我国拥有庞大的人才储备,各大学及研究机构每年都会输送大批生命科学领域的新生人才,各大生物研发实验室也在不断培养技术骨干,众多国际知名华裔科学家也都渴望归国效力。如果能够将生物技术药物的研发中心、生产基地引进我国,形成真正的生物技术药品创新中心,则可推动持续创新,促进生物技术药物研发和相关人才的培养。
[1] U.S.food and drug administration.Orphan Drug Designations and Approvals Database[EB/OL].[2015-03-08]http://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm.
[2] 易八贤,王广平,姬海红,等.美国孤儿药法案30年历程与我国新药创新制度体系完善[J].中国新药杂志,2014(10):1107-1114.
[3] 斯科特.制度与组织——思想观念与物质利益[M].姚伟,王黎芳,译.北京:中国人民大学出版社,2010:56-59.
[4] HOFFMAN A J.Institutional Evolution and Change:Environmentalism and the US Chemical Industry[J].Academy of Management Journal,1999,42(4):351-371.
[5] 田志龙,张泳.中国电力行业的演变:基于制度理论的分析[J].管理世界,2002(12):69-76.
[6] BERGER P L,LUCKMANN T.The Social Construction of Reality:A Treatise in the Sociology of Knowledge[M].New York:Doubleday Anchor,1966:103-105.
[7] GIORGI,A,GIORGI,B.Qualitative Psychology:A PracticalGuide to Research Methods[M].Thousand Oaks:Sage Publications,2003:53-80.
[8] DUNKLE M.30-year Retrospective:National Organization for Rare Disorders,the Orphan Drug Act,and the Role of Rare Disease Patient Advocacy Groups[J].Orphan Drugs:Research and Review,2014(4):19-27.
[9] 刘炳林.国外孤儿药政策概述及启示[J].中药新药与临床药理,2002,13(5):332-333.
[10] ORPHANET.About rare diseases.[EB/OL].[2013-11-22].http://www.orpha.net/consor/cgi-bin/Education_AboutRareDiseases.php?lng=EN.
[11] U.S.food and drug administration.How to Apply for Orphan DrugDesignation[EB/OL].[2013-06-12].http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/HowtoapplyforOrphanProduct-Designation/ucm365086.htm.
[12] EVALUATEPHARMA.Orphan Drug Report2014[EB/OL].[2015-04-13].http://info.evaluategroup.com/rs/evaluatepharmaltd/images/2014OD.pdf.
[13] WAXMAN H.The Waxman Report:How Congress Really Works[M].New York:Grand Central Publishing,2009:110-132.
[14] 田春英.解读特色企业——美国健赞公司[J].中国医药生物技术,2010(3):238-239.
[15] 李天柱,银路,程跃.美国领先生物制药企业的成功要素及其启示[J].管理学报,2010,7(9):1335-1342.
(责任编辑:冯 蓉)
Institutional Change of Orphan Drug Act and Im p lications for China Based on Study of Chinese Biopharmaceutical Industry
GAO Shanxing,HAN Chen
(School of Management,Xi′an Jiaotong University,Xi′an 710049,China)
Formation and thirty-year institutional change of Orphan Drug Act are discussed usingmodern institutional theory and phenomenon analysis,with a specific focus on the legislation adjustments and the impact of institutional environment on industry development.Results show that institutional change of Orphan Drug Act is a three-stage societal process and appropriate institutional environments will significantly enhance biopharmaceutical industry development.Critical agents and institutional pillars are different across stages.Patients,federal government,and enterprises are playing a dominant role in the three stages respectively.Cultural-cognitive,regulative,and normative pillars promote institutional change respectively.Finally,the implication for Chinese biopharmaceutical industry is to continue building appropriate institutional environments.Specifically,Chinese government is urged to improve the regulatory pillar to exert its legislative influence,to strengthen the cultural-cognitive pillar to accumulatemore,and to reinforce the normative pillar to achieve benign progress.
the United States;Orphan Drug Act;institutional theory;phenomenon analysis;institutional change;Chinese biopharmaceutical industry
D904.6
D912.182
1008-245X(2015)06-0100-07
10.15896/j.xjtuskxb.201506017
2015-03-30
国家社会科学基金重大项目(11&ZD170);中央高校基本科研业务费专项项目
高山行(1963- ),男,西安交通大学管理学院教授,博士生导师。
