Design and evaluation of nicorandil extended-release tablet
2015-05-16Ju-YoungKim,Chun-WoongPark,Beom-JinLee等
Original Research Paper
Design and evaluation of nicorandil extended-release tablet
Ju-Young Kima,Chun-Woong Parkb,Beom-Jin Leec,Eun-Seok Parkd, Yun-Seok Rheee,*
aCollege of Pharmacy,Woosuk University,Wanju-gun 565-701,Republic of Korea bCollege of Pharmacy,Chungbuk National University,Cheongju 361-763,Republic of Korea cCollege of Pharmacy,Ajou University,Suwon 443-749,Republic of Korea dSchool of Pharmacy,Sungkyunkwan University,Suwon 440-746,Republic of Korea eCollege of Pharmacy and Research Institute of Pharmaceutical Sciences,Gyeongsang National University, Jinju 660-701,Republic of Korea
ARTICLEINFO
Article history:
Received 20 June 2014
Received in revised form
11 September 2014
Accepted 12 September 2014
Available online 22 September 2014
Nicorandil
A
The aim of this study was to design and evaluate extended-release formulations of a model drug,nicorandil,in order to achieve the desired steady-state plasma concentration of drug in vivo.Simulation was employed to estimate optimum dissolution and absorption rate of nicorandil.The dissolution test was employed using pH 1.2,4.0,6.8 buffer solution,or water,to measure the in vitro release behaviors of nicorandil formulations.A single dose (15 mg)of each formulation was orally administered to four beagle dogs under fasted conditions,and the pharmacokinetic parameters were calculated.The in vitro/in vivo relationship of the extended-release formulation was con fi rmed using in vitro dissolution pro fi les and plasma concentrations of drug in beagle dogs.Nicorandil was released completely within 30 min from the immediate-release tablets and released for 24 h from the extended-release tablets.The nicorandil plasma concentration could be modi fi ed by adjusting the drug release rate from the extended-release formulation.The release rate of nicorandil was the rate-limiting step in the overall absorption of drug from the extendedrelease formulations.These results highlight the potential of a nicorandil extended-release formulation in the treatment of angina pectoris.
©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
1. Introduction
Nicorandil(Fig.1)is a vasodilator acting through an increase of both membrane potassium ion conductance and intracellular cGMP concentration in vascular smooth muscle[1].It is clinically used in the treatment of angina pectoris.Nicorandil is not metabolized signi fi cantly by the liver during passage through the portal system(lack of fi rst-pass effect).Thus,it easily enters the systemic blood fl ow,resulting in almost complete bioavailability.After oral administration of a 5-,10-, 20-,or 40-mg dose,there is a linear relationship between the doses and increases of maximum plasma concentrations and area under the curve,demonstrating that the pharmacokinetics of nicorandil are linear[2,3].Because of its short elimination half-life(1 h),the drug has to be given frequently at 5 mg immediate-release(IR)tablet three times a day.To reduce the frequency of administration and to improve patient compliance,a once-daily extended-release(XR)formulation of nicorandil is desirable.Moreover,the conventional therapy may result in high fl uctuation in plasma concentration of the drug resulting in unwanted side effects[4].Hence, the development of XR formulations for nicorandil that could providethedesiredconstantdrugdeliveryforapredetermined period is bene fi cial for an effective and safe therapy of angina pectoris.An essential step in developing XR formulation is to accommodate both the in vitro and in vivo properties of the drug.Here,we compared a novel,XR formulation of nicorandil with an IR formulation,both in vitro and in vivo.
The aims of this study were to design XR formulations of nicorandil and to evaluate the possibility of XR formulations of nicorandil for fi nding its ability in providing the desired steady-state plasma concentration of drug in vivo.Simulation was employed to estimate optimum dissolution and absorption rate of nicorandil.The in vitro/in vivo relationship of XR formulation was con fi rmed using in vitro dissolution pro fi les and plasma concentrations after a single dose oral administration under fasting condition in beagle dogs.
2. Materials and methods
2.1. Materials

?
The following materials were used as received without further puri fi cation:nicorandil was purchased from Jiangsu Tasly Diyi Pharm.Co.,Ltd.(Jiangsu,China).Analytical grade solvents such as acetonitrile,isopropyl alcohol and ethyl acetate were supplied by Merck&Co.(Darmstadt,Germany).Potassium dihydrogen phosphate,and dibasic sodium phosphate(Shinyo Pure Chemicals Co.,Japan)were of analytical grade.Other excipients were of reagent grade.Deionized water was purifi ed using a Milli-Q system(Millipore,Milford,MA,USA).
2.2. Pharmacokinetic modeling and simulation of oral nicorandil
Exploitation of the new XR formulation with made-to-order release characteristics for nicorandil the optimum dissolution rate to maintain therapeutic plasma concentration was computed employing MicroMath®Scientist(MicroMath Scienti fi c Software,Salt Lake City,UT,USA).Pharmacokinetic parameters reported in the literature were used to predict the pharmacokinetic pro fi les of nicorandil after oral administration[3].The intrinsic absorption rate constant,ka,was systematically varied from 0.1 to 3 h-1.The optimum pro fi le of dissolution was calculated by Wagner-Nelson method with the simulated plasma concentration data[5,6].
2.3. Preparation of nicorandil extended-release tablets
Nicorandil XR tablets were prepared by a direct compression method.Table 1 lists the composition of the fi nal formulation. Brie fl y described,nicorandil bulk powder was mixed carefully with low substituted hydroxypropyl cellulose,microcrystalline cellulose,hypromellose,silicon dioxide,and magnesium stearate in sequence.The mixture was compressed into tablets with a weight of 200 mg by a MINI Press II SF tablet machine(Karnavati Engineering Ltd.,Gujarat,India).Each tablet contained 7.5 mg of nicorandil.
2.4. In vitro release study
The in vitro dissolution tests were performed using USP dissolution apparatus 2(DST-810,Lab fi ne Inc.,Korea).One IR tablet(Sigmat®5 mg,JW Pharm.Co.,Korea)corresponding to 5 mg of nicorandil or one XR tablet corresponding to 7.5 mg of nicorandil was placed in a vessel with 900 ml of the dissolution medium at 37±0.5°C rotating at 50 rpm.The dissolution media were simulated gastric fl uid(pH 1.2)without pepsin, acetate buffer solution(pH 4.0),phosphate buffer solution (pH 6.8)and deionized water.Aliquots were removed periodically and assayed for nicorandil by high performance liquid chromatography(HPLC)as described in drug analysis section.Each dissolution test was repeated 6 times and the mean values with standard deviation are presented.
2.5. In vivo release study
A cohort of four healthy male beagle dogs(Marshall Beijing, China)(10-12 kg)were used under fasted conditions for 18 h.The dogs had free access to water.Three Sigmat®5 mg (JW Pharm.Co.,Korea)tablets(5 mg×3)or two nicorandil XR(7.5 mg×2)tablets corresponding to 15 mg of nicorandil were orally administered to four male beagle dogs,with a washout period of at least 1 week between two consecutive administrations.Serial blood samples(3 ml each)were collected from a forearm vein in a heparinized tube at predetermined times extending to 24 h.Plasma was immediately obtained by centrifuging blood samples at 3000 rpm for 10 min(Hanil micro 12,Hanil,Korea).All the samples were stored frozen at-70°C until analysis.Plasma levels of nicorandil were assayed by HPLC as described in drug analysis section.
2.6. Analysis of nicorandil using HPLC
Analysis of nicorandil was performed using an HPLC method [7,8].The HPLC procedures were fully validated prior to their routine use,with the area under the peak values used for the calculations.The validation tests included system suitability, accuracy,reproducibility,linearity and ruggedness.
The HPLC system(Waters,Milford,MA,USA)consisted of Waters Alliance 2690 HPLC pump with a Waters Alliance 2690 autosampler and column oven,a Waters 996 photo diode array detector set at 254 nm,and an HPLC system manager (Waters,Millennium).The column used was a Zorbax®Phenyl column(150 mm×4.6 mm i.d.;5 μm;Phenomenex,CA,USA) operated at room temperature.The analytical mobile phase, consisting of acetonitrile-isopropyl alcohol-water(12:2:86,v/ v),was continuously passed through the analytical column at a fl ow rate of 1.0 ml/min.A portion(160 μl)of the sample was injected into the column.
The plasma concentrations of nicorandil were determined using a validated HPLC method[7,8].Brie fl y,to the plasma sample containing internal standard,one drop of 0.1M NaOH was added,followed by 1.0 ml of ethyl acetate.The mixture was vortexedfor 30 s,and centrifuged for 10 minat 2000 g.The upper ethyl acetate layer was removed by Pasteur pipet and transferred to test tube.The extraction procedure was repeated,and the organic layers were combined and evaporated under nitrogen.The residue was reconstituted with 200 μl of the mobile phase on a vortex mixer for 60 s.The reconstituted solution was then assayed.
2.7. In vivo data analysis
The pharmacokinetic parameters were calculated through a weighted least squares procedure,with the aid of the nonlinear regression programs,SigmaPlot ver.11.0(SPSS Inc., Chicago,IL,USA)and MicroMath®Scientist.Plasma drug concentrations for 12 h were used to calculate pharmacokinetic parameters because plasma drug concentrations at 24 h were less than limit of quantitation.
AUC0-12is the area under the plasma concentration versus time curve,calculated using the trapezoidal rule for the time interval 0 to the last measurable point,12 h.The total areas under plasma level curves,AUCinf,were calculated by combining the areas from 0 to 12 h,estimated by the trapezoidal rule,with those obtained from 12 h to infi nity,calculated as the ratio C12/ke,in which C12is the corrected plasma level at 12 h and keis the elimination rate constant[9].Elimination constant(ke)was estimated by fi tting the logarithm of the concentrations versus time to a straight line over the observed exponential decline.Elimination half-life,t1/2was calculated as follows:t1/2=0.693/ke. The peak plasma concentration(Cmax)and time to reach the maximum drug plasma concentration(Tmax)were determined from visual inspection of the concentration-time plots.
The Wagner-Nelson method[5,6]was used to calculate the percentage of the drug absorbed:
where F(t)is the amount absorbed.The percent absorbed is determined by dividing the amount absorbed at any time by the plateau value,keAUCinfand multiplying this ratio by 100:
2.8. In vitro-in vivo relationship
Analysis of in vitro-in vivo relationships was of the form[10,11]: whereFais thefractionof thetotal amount of drugabsorbedat time t,fais the fraction of the dose absorbed at t=∞,α is the ratio of the fi rst-order permeation rate constant(kp)to the fi rst-order dissolution rate constant(kd),and Fdis the fraction of drug dose dissolved at time t.Fawas determined by the Wagner-Nelson method from the plasma pro fi les,and Fdwas obtained from the dissolution pro fi les.Eq.(3)was fi tted to the Favs.Fddata to yield an estimate of α.The fi rst-order permeation rate constant(kp)was calculated from kp=α kd. All regressions employed non-linear least squares by the MicroMath®Scientist(MicroMath Scienti fi c Software,Salt Lake City,UT,USA).
2.9. Statistics
Statistical comparisons of pharmacokinetic parameters were performed with the Student's t-test.Statistical signi fi cance was accepted for P<0.05.
3. Result and discussion
3.1. Pharmacokinetic modeling and simulation of oral nicorandil
In this study,we assumed the drug release rate is identical to the drug absorption rate because the drug is rapidly andalmost completely absorbed from the gastrointestinal tract [3].Therefore,the pattern of nicorandil plasma concentrations could be modi fi ed by adjusting the drug release rate of XR tablets.The optimum absorption rate of simulated plasma concentration was found to be ka=0.2 h-1for the XR nicorandil providing more than 90%drug release maintained over the period of 24 h.Utilizing the calculated absorption rate, 0.2 h-1,the optimum dissolution pro fi le was simulated by Wagner-Nelson method.
3.2. In vitro release study
To measure release rates of drug,the in vitro dissolution tests of nicorandil tablets were performed in dissolution media: water,pH 1.2,4.0 and 6.8 buffer solutions(Fig.2).Nicorandil was quickly released from the IR tablets,whereas the XR tablet showed slow release for 24 h.Within 30 min,95%of drug was released from the IR tablet in all dissolution media (data not shown),but the time for 80%drug release from the XR tablet was about 8 h(Fig.2).Dissolution rates of the extended-release tablets were almost unaffected by pH.As shown in Fig.2,dissolution pro fi les of XR tablets in all dissolution media showed similar extended-release pattern to the target dissolution pro fi le(ka=0.2).
3.3. In vivo release study
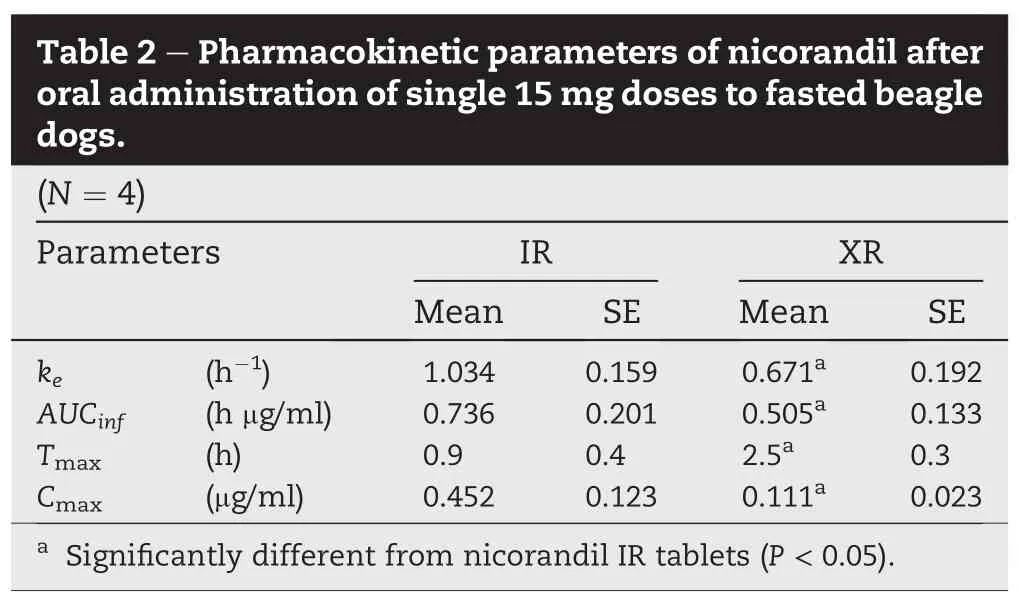
Plasma drug concentration vs.time pro fi les of oral nicorandil in beagle dogs are shown in Fig.3.In vivo release study revealed that XR formulations exhibited an extended-release pattern for a period of 12 h.Nicorandil IR formulations showed rapid drug release and absorption within 1 h and fast drug elimination pro fi le for a period of 6 h.Pharmacokinetic parameters for the two formulations are listed in Table 2.The pharmacokinetic parameters of nicorandil XR tablet were signi fi cantly(P<0.05)different from those obtained with IR tablet.It took 2.5±0.3 h(Tmax)to reach maximum concentration of 0.111±0.023 μg/ml(Cmax)from XR tablets.However,the Cmax(0.452±0.123 μg/ml)of the drug reached within 0.9±0.4 h and declined rapidly from IR tablets.The elimination rate constant of nicorandil from XR tablet was signi fi cantly decreased in comparison with IR tablet(P<0.05; 0.671 h-1vs.1.034 h-1)and the AUCinfof XR tablet was also signi fi cantly decreased in comparison with IR tablet(P<0.05). As for XR formulation of drug product,most of XR formulation is commonly administered with food to get enough gastric emptying time.In this study,the comparative pharmacokinetic study was conducted under fasting state with dogs.In case of dogs,gastrointestinal tract is shorter than that of human,and therefore,XR formulation have just 60-70%of bioavailability compared to IR formulation especially for fasting state study.From the results,it was suggested that the pattern of nicorandil plasma concentrations could be modi fi ed by adjusting the drug release rate from XR tablets.
3.4. In vitro-in vivo relationship
In this study,Faand Fddata from two nicorandil tablet formulations are analyzed.Neither a linear nor a linearized relation between Faand Fdis assumed.The main intent of this study was to make use of the in vitro-in vivo relationship for each tablet formulation in order to elucidate the relative roles of dissolution and intestinal permeation in overall nicorandil absorption from each dosage form[11].
Fig.4 plots the fraction absorbed(Fa)versus the fraction dissolved(Fd)and graphs the mean fi t of Eq.(3)to each of the two tablet formulations.Each pro fi le represents the in vitroin vivo relationship for each respective product.The curve characterizing the dissolution and absorption of nicorandil from the IR tablet rapidly moves from(Fa=0,Fd=0)in the lower left corner of the phase plane at t=0 to the 15 min data point in the lower right corner of the phase plane.This portion of the curve demonstrates the rapid dissolution of nicorandil during the fi rst 15 min and the relatively low fraction of absorbed drug at 15 min.Over the following 15 min,the curve“upward”due to the relatively large fraction in nicorandil absorption while only the small remaining fraction of nicorandil dissolves during that time frame.For the next 30 min, essentiallyonly nicorandil absorptionoccurssincedissolution had been complete.Unlike for IR formulation,the relationship between Faand Fdfor XR formulation is substantially linear (Fig.4).The upward trajectory characterizing the dissolution and absorption of nicorandil from the XR tablet gradually and linearly moves from(Fa=0,Fd=0)in the lower left corner of the phase plane at t=0 to the data point in the higher right corner of the phase plane.This pattern of the trajectory refl ects the slow dissolution of nicorandil during the 12 h but the rapid drug absorption after drug released from XR tablets.It is shownthattheabsorptionofthe drugtakes placeat nearlythe same time of the release of the drug over the whole time frame.For the 12 h,essentially nicorandil absorption occurs since dissolution had been continued.Therefore this slowly dissolving nicorandil XR formulation appears to be substantially dissolution rate limited.Table 3 summarizes the mean (±SE)values for α,kdand kp.For IR formulation,α was less than 1(α≪1),which indicates that kd≫kp.However it may be dif fi cult to determine the rate-limiting step because the dissolution rate of drug from IR formulations was too fast.For XR formulation,α was larger than 1(α≫1)and this indicates that dissolution,rather than intestinal permeation,was the rate-limiting step in the overall absorption of drug.

4. Conclusion
The present study demonstrates that an XR formulation of nicorandil was successfully designed and developed.The nicorandilplasmaconcentration could bemodi fi ed by adjustingthe drugrelease ratefrom XR formulation.Intestinal permeation of nicorandil,rather than dissolution,was the rate-limiting step in the overall absorption of drug from the IR formulation.However release rate of nicorandil from XR formulation was the rate-limiting step in the overall absorption of drug.Moreover,these results highlight the potential of a nicorandil XR formulation for an effective and safe therapy of angina pectoris.
Acknowledgement
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF)fundedby theKorean Ministryof Education,Science and Technology(NRF-2012R1A1A1013210)and by a grant of the Korean Health Technology R&D Project,Ministry of Health, Welfare&Family Affairs,Republic of Korea(A092018).
REFERENCES
[1]Ishizaki T,Chiba K,Suganuma T,et al.Pharmacokinetics of nicorandil,a new coronary vasodilator,in dogs.J Pharm Sci 1984;73:494-498.
[2]Frydman AM,Chapelle P,Diekmann H,et al. Pharmacokinetics of nicorandil.Am J Cardiol 1989;63:25J-33J.
[3]Frydman A.Pharmacokinetic pro fi le of nicorandil in humans:an overview.J Cardiovasc Pharmacol 1992;20(Suppl. 3):S34-44.
[4]Krishnaiah YS,Al-Saidan SM,Chandrasekhar DV,et al. Bioavailability of nerodilol-based transdermal therapeutic system of nicorandil in human volunteers.J Control Release 2005;106:111-122.
[5]Wagner JG,Nelson E.Percent absorbed time plots derived from blood level and/or urinary excretion data.J Pharm Sci 1963;52:610-611.
[6]Wagner JG,Nelson E.Kinetic analysis of blood levels and urinary excretion in the absorptive phase after single doses of drug.J Pharm Sci 1964;53:1392-1403.
[7]Bachert EL,Fung HL.High-performance liquid chromatographic method for stability and pharmacokinetic studies of nicorandil.J Chromatogr 1993;619:336-341.
[8]Schwende FJ,Lewis RC.Determination of nicorandil in plasma using high-performance liquid chromatography with photoconductivity and ultraviolet detection.Application to pre-clinical pharmacokinetics in beagle dogs.J Chromatogr 1990;525:151-160.
[9]Wagner JG.Pharmacokinetic absorption plots from oral data alone or oral/intravenous data and an exact Loo-Riegelman equation.J Pharm Sci 1993;72:838-842.
[10]Polli JE,Crison JR,Amidon GL.Novel approach to the analysis of in vitro-in vivo relationships.J Pharm Sci 1996;85:753-760.
[11]Polli JE.In vitro-in vivo relationships of several“immediate”release tablets containing a low permeability drug.Adv Exp Med Biol 1997;423:191-198.
*Corresponding author.College of Pharmacy and Research Institute of Pharmaceutical Sciences,Gyeongsang National University,Jinju, Gyeongsangnam-do 660-701,Republic of Korea.
E-mail addresses:ysrhee@gnu.ac.kr,atomicos@naver.com(Y.-S.Rhee).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2014.09.003
1818-0876/©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
In vitro
In vivo
Pharmacokinetic Extended-release
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- A review on phospholipids and their main applications in drug delivery systems
- Studies on thermoresponsive polymers:Phase behaviour,drug delivery and biomedical applications
- Development and evaluation of vinpocetine inclusion complex for brain targeting
- A new self-emulsifying formulation of mefenamic acid with enhanced drug dissolution
- Can semipermeable membranes coating materials in fl uence in vivo performance for paliperidone tri-layer ascending release osmotic pump tablet: In vitro evaluation and in vivo pharmacokinetics study
- Mycosynthesis,characterization and antibacterial properties of AgNPs against multidrug resistant (MDR)bacterial pathogens of female infertility cases