端叠氮基超支化聚酯的制备与优化
2015-05-10姚大虎白森虎柴春鹏罗运军
王 晓, 姚大虎, 白森虎, 柴春鹏, 罗运军
(1. 河南科技大学化工与制药学院,河南 洛阳 471023; 2. 黎明化工研究设计院有限责任公司, 河南 洛阳 471001; 3. 北京理工大学材料学院, 北京 100081)
1 引 言
随着以叠氮缩水甘油醚(GAP)、3-甲基-3-叠氮甲基氧丁环(AMMO)、3,3-双(叠氮甲基)氧丁环(BAMO)的均聚物与共聚物为代表的叠氮含能粘合剂的广泛应用,不同结构的叠氮齐聚物增塑剂研究也受到了广泛关注[1-2]。叠氮齐聚物增塑剂在解决了其他含能增塑剂与叠氮粘合剂间因含能基团不同而引起的相容性问题的同时,也解决了小分子叠氮增塑剂在使用中普遍存在的易迁移、易挥发等缺点,成为含能增塑剂研究领域中的一个重要研究方向。
文献最早报道的叠氮齐聚物增塑剂是由端羟基聚环氧氯丙烷(PECH)与叠氮化钠反应制备得到的端羟基GAP齐聚物[3-4],因其具有与聚醚或聚酯预聚体相同的端羟基,固化时与二异氰酸酯反应,所以,对其端羟基进行酯化改性得到了端酯基GAP齐聚物(GAPE)[5],对其端羟基进行叠氮化改性得到了端叠氮基GAP齐聚物(GAPA)[6-8],对其端羟基同时进行酯化和叠氮化改性得到了端酯基端叠氮基GAP齐聚物(GAPAE)[9]。为得到结构可控的GAP齐聚物,王晓等人在端羟基聚环氧氯丙烷的制备过程中,以不同的多元醇为起始剂进行阳离子开环聚合,而后端羟基被酯化[10]或先磺酰化后叠氮化取代[11],分别得到两类端基不同的结构可控的多臂型GAP齐聚物。为充分利用树形分子准球形结构具有的低玻璃化转变温度和低粘度的特性,Pant C S等人[12-13]制备出端叠氮基树形聚酯增塑剂,他们以三羟甲基丙烷或季戊四醇为中心核,经过二羟甲基丙酸(DMPA)羟基酯化保护、端酯基树形聚酯合成、端羟基树形聚酯合成、端磺酰氧基树形聚酯合成、端叠氮基树形聚酯合成等5个步骤共96h制得端叠氮基树形聚酯增塑剂。为解决端叠氮基树形聚酯制备过程的反应步骤多、反应周期长、成本高等问题,考虑到超支化聚合物增塑聚氨酯粘合剂后形成互穿聚合物网络结构可同时提高聚氨酯体系的拉伸强度和延伸率,王晓等人[14]以超支化聚酯结构代替树形聚酯结构,以端羟基超支化聚酯为基体,端羟基经磺酰化后得到端磺酰氧基超支化聚酯(HBPE-OTS)、而后端磺酰氧基被叠氮化后得到端叠氮基超支化聚酯(HBPE-N3),将合成步骤简化为3步,降低了产品成本。本研究在文献[14]的基础上,从反应介质、反应温度、反应时间、催化剂用量等角度对HBPE-OTS端基叠氮化取代制备HBPE-N3的反应进行优化研究。
2 实验部分
2.1 主要原料与仪器
HBPE-OTS,实验室自制(采用文献[14]中的方法,以三羟甲基丙烷为核,采用准一步法制备得到端羟基超支化聚酯,而后在吡啶中与对甲苯磺酰氯反应制备); 二甲基亚砜(DMSO),AR,天津市永大化学试剂开发中心; 二甲基甲酰胺,AR,西陇化工股份有限公司;N-甲基吡咯烷酮,AR,上海埃彼化学试剂有限公司; 吡啶,AR,天津市化学试剂有限公司; 叠氮化钠,AR,郑州派尼化学试剂有限公司; 四丁基溴化铵,AR,天津市光复精细化工研究所; 二氯甲烷,AR,北京化工厂。上述除吡啶、二甲基亚砜在使用前需加入4A分子筛干燥24 h外,其他试剂无需进一步处理。
傅里叶变换红外光谱仪,Nicolet 8700型,Thermo公司; 真空旋转蒸发仪,YRE201D型,予华公司。
2.2 实验步骤[14]
向装有机械搅拌器、温度计、回流冷凝管和氮气入口的四口圆底烧瓶中依次加入DMSO、HBPE-OTS和四丁基溴化铵催化剂,通氮气,开启搅拌,用油浴控制升温至95 ℃,待上述物料混合均匀后分批加入叠氮化钠,加料结束后恒温反应24 h。在此反应中,HBPE-OTS中的端磺酰氧基被叠氮基所取代,生成端叠氮基超支化聚酯,反应路线见Scheme 1。

Scheme 1 Synthetic route of azide-terminated hyperbranched polyesters(HBPE-N3)
反应停止后, 加入约2倍反应混合液体积的蒸馏水洗涤,有棕红色粘稠状沉淀生成,然后加入二氯甲烷,粘稠状沉淀溶解。分离出上层水相后,用蒸馏水洗涤下层有机相3次,分离得到的有机相在90 ℃下真空旋蒸约2 h,得黄红色透明油状液体,即为端叠氮基超支化聚酯。
2.3 反应程度表征
HBPE-OTS中端磺酰氧基的反应程度(叠氮化率)f可用于表征HBPE-N3的合成情况,采用N元素分析法或定量红外光谱法[15]进行表征。对多个样品两种表征方法得到的反应程度进行比较研究,发现两种表征手段得到的反应程度基本一致。采用N元素分析法时,不同反应程度产物的N含量可根据理论计算得到,用元素分析仪对不同反应条件下产物进行元素分析,根据N含量表征结果得到产物相对应的反应程度。采用定量红外光谱方法测定时,以超支化聚酯结构中的甲基特征吸收峰为内标峰,设反应前原料HBPE-OTS中端磺酰氧基特征吸收峰强度与甲基特征吸收强度之比为A,反应结束后产物中的端磺酰氧基特征吸收峰强度与甲基特征吸收强度之比为B,则反应程度(叠氮化率)f=(1-B/A)×100%。与N元素分析法相比较,定量红外光谱法表征f较为便捷,故本研究采用定量红外光谱法。
3 结果与讨论
3.1 反应介质对HBPE-N3合成的影响
叠氮化反应常用的反应介质有二甲基亚砜(DMSO)、二甲基甲酰胺(DMF)和N-甲基吡咯烷酮(NMP)[4,7-8,16-17]。在反应温度95 ℃、反应时间24 h、叠氮化钠的加入量为化学反应量的1.5倍、四丁基溴化铵(催化剂)的加入量为叠氮化钠物质的量的5.0%的条件下,考察了上述三种反应介质对HBPE-OTS的端磺酰氧基叠氮化取代反应的影响,结果如表1所示。
表1 反应介质对HBPE-OTS反应程度的影响
Table 1 Influence of reaction medium on reaction extent of HBPE-OTS

reactionmediumDMSODMFNMPf/%99.895.297.3
Note:fis reaction extent.
由表1可知,三种反应介质中,HBPE-OTS的反应程度均超过了95%,但是,DMSO为反应介质时的反应程度最高,HBPE-OTS的端磺酰氧基几乎全部被叠氮基团所取代,这与HBPE-OTS和HBPE-N3在DMSO中的溶解性较好有关。三种反应介质中,DMF为反应介质时的反应程度最低,这可能与DMF在高温下长时间反应容易分解有关,其分解副产物影响了HBPE-OTS的叠氮化取代反应。NMP价格相对较高,成本较高,并且其作为反应介质时的反应程度稍低于DMSO。综合叠氮化反应程度、反应成本等因素,HBPE-OTS的端磺酰氧基叠氮化取代反应的较佳反应介质为DMSO。
3.2 反应温度对HBPE-N3合成的影响
以DMSO为反应介质,反应时间24 h,叠氮化钠加入量为化学反应量的1.5倍,催化剂四丁基溴化铵的加入量为叠氮化钠物质的量的5.0%,考察了反应温度对HBPE-OTS的端磺酰氧基叠氮化取代反应的影响,结果如表2所示。
表2 反应温度对HBPE-OTS反应程度的影响
Table 2 Influence of reaction temperature on reaction extent of HBPE-OTS

T/℃658095110f/%87.192.399.897.3
由表2可知,在95 ℃以下时,随着反应温度升高,HBPE-OTS的端磺酰氧基叠氮化取代反应程度也逐渐提高(在95 ℃时的反应程度最高,为99.8%),产品HBPE-N3为红褐色透明粘稠状液体。但是,当温度升高至110 ℃后,相同条件下HBPE-OTS的端磺酰氧基叠氮化取代反应程度下降至97.3%,并且产品为黑红色不透明粘稠状液体。这可能是DMSO和少量HBPE-N3产品在较高温度下发生副反应的缘故。同时考虑到在较高反应温度下HBPE-OTS的端磺酰氧基叠氮化取代反应存在安全性问题,HBPE-OTS的端磺酰氧基叠氮化取代反应的较佳反应温度为95 ℃。
3.3 反应时间对HBPE-N3合成的影响
以DMSO为反应介质,反应温度95 ℃,叠氮化钠加入量为化学反应量的1.5倍,催化剂四丁基溴化铵为叠氮化钠物质的量的5.0%,考察了不同反应时间对HBPE-OTS的端磺酰氧基叠氮化取代反应的影响,结果如图1所示。
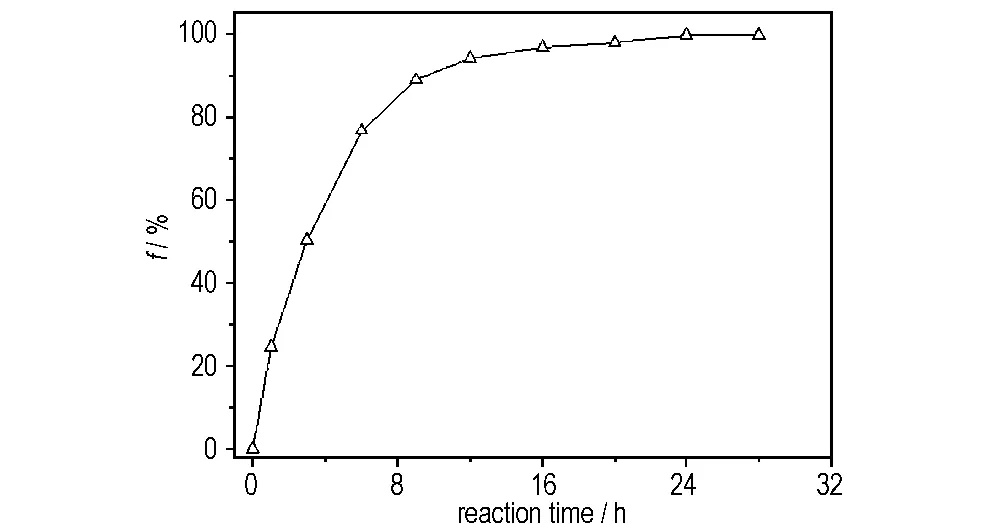
图1 反应时间对HBPE-OTS反应程度的影响
Fig.1 Influence of reaction time on reaction extent of HBPE-OTS
由图1可知,随着反应时间的延长,HBPE-OTS的端磺酰氧基叠氮化取代反应的反应程度不断提高。在反应初期,叠氮化取代反应进行较快,12h内约有94.0%的端磺酰氧基被叠氮基取代,随后反应速率减缓,24 h时叠氮化取代反应达99.8%,此后叠氮化取代反应程度不再随着时间的增加而提高,可以认为此时叠氮化取代反应进行完毕。因此,HBPE-OTS的端磺酰氧基叠氮化取代反应的较佳反应时间为24 h。
3.4 叠氮化试剂过量度对HBPE-N3合成的影响
以DMSO为反应介质,反应温度95 ℃,反应时间24 h,催化剂四丁基溴化铵加入量为叠氮化钠物质的量的5.0%条件下,考察了不同叠氮化钠过量度(加料量/化学反应量-1)对HBPE-OTS的端磺酰氧基叠氮化取代反应的影响,结果如表3所示。
表3 叠氮化钠过量度对HBPE-OTS反应程度的影响
Table 3 Influence of excessive degree of sodium azide on reaction extent of HBPE-OTS

excessivedegree/%10305080f/%88.394.599.899.8
由表3可知,在其他反应条件相同的情况下,HBPE-OTS的端磺酰氧基叠氮化取代反应程度随着叠氮化钠过量度增加而逐渐提高,但是,当叠氮化钠过量度增加到50%后,HBPE-OTS的端磺酰氧基叠氮化取代反应程度不再随着叠氮化钠过量度的增加而提高。因此,HBPE-OTS的端磺酰氧基叠氮化取代反应时叠氮化钠的较佳过量度为50%,也就是说,叠氮化钠加入量为化学反应量的1.5倍最佳。
3.5 催化剂四丁基溴化铵加入量对HBPE-N3合成的影响
以DMSO为反应介质,反应温度95 ℃,反应时间24 h,叠氮化钠加入量为化学反应量的1.5倍,考察了催化剂四丁基溴化铵不同加入量(催化剂与叠氮化钠物质的量的比)对HBPE-OTS的端磺酰氧基叠氮化取代反应的影响,实验结果如表4所示。
表4 四丁基溴化铵加入量对HBPE-OTS反应程度的影响
Table 4 Influence of additive amount of tetrabutylammonium bromide on reaction extent of HBPE-OTS

additiveamount/%1.03.05.07.0f/%89.295.199.899.8
由表4可知,在其他反应条件相同的情况下,HBPE-OTS的端磺酰氧基叠氮化取代反应程度随着催化剂的加入量增加而逐渐提高,但是,当超过5.0%后,HBPE-OTS端磺酰氧基的叠氮化取代反应程度不再随着催化剂加入量的增加而提高。因此,HBPE-OTS的端磺酰氧基叠氮化取代反应时催化剂的较佳加入量为叠氮化钠物质的量的5.0%。
4 结 论
以端磺酰氧基超支化聚酯为前驱体,在二甲基亚砜中,与叠氮化钠反应,其端磺酰氧基被叠氮基所取代,制备得到目标产品端叠氮基超支化聚酯(HBPE-N3)。
端磺酰氧基超支化聚酯叠氮化取代反应的优化条件: 以DMSO为反应介质,反应温度95 ℃,反应时间24 h,叠氮化钠加入量为化学反应量的1.5倍,催化剂四丁基溴化铵加入量为叠氮化钠物质的量的5.0%。在此条件下,99.8%的端磺酰氧基被叠氮基所取代。
参考文献:
[1] 王连心,牛群钊,崔小军,等.齐聚物含能增塑剂研究进展[J].化学推进剂与高分子材料,2009, 7(6): 23 -25.
WANG Lian-xin, NIU Qun-zhao, CUI Xiao-jun, et al. Research progress of energetic oligomer plasticizers[J].ChemicalPropellants&PolymericMaterials, 2009, 7(6): 23 - 25.
[2] 葛震, 罗运军, 郭凯, 等. BAMO 均聚物及共聚物合成研究进展[J]. 含能材料,2009, 17(6): 745-750.
GE Zhen, LUO Yun-jun, GUO Kai, et al. Review on synthesis of BAMO homopolymer and copolymers[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2009, 17(6): 745-750.
[3] Ahad E. Direct conversion of epichlorohydrin to glycidyl azide polymer: United States, 4891438[P]. 1990-01-02.
[4] Flanagan J E, Frankel M B. Energetic hydroxy-terminated azido polymer: United States, 4268450[P]. 1981-05-19.
[5] Flanagan J E, Wilson E R. Glycidyl azide polymer esters: United States, 4938812[P]. 1990-07-03.
[6] Ampleman G. Synthesis of a diazido terminated energetic plasticizer: United States, 5124463[P]. 1992-06-23.
[7] Milton B F, Cunningham M A, Wilson E R. Glycidyl azide polymer azide: United States, 2285624[P]. 1995-07-19.
[8] Wilson E R, Frankel M B. Azide-terminated azido compound: United States, 4781861[P]. 1988-11-01.
[9] Bouchez J, Graindorge H, Soriaux C. Polyglycidyl azides comprising an acyloxy terminal group and an azide terminal group: United States, 6323352[P]. 2001-11-27.
[10] 王晓, 罗运军, 葛震, 等. 多臂型端酯基聚叠氮缩水甘油醚的合成与表征[J]. 高分子材料科学与工程,2012, 28(2): 1-4.
WANG Xiao, LUO Yun-jun, GE Zhen, et al. Synthesis and characterization of Multi-armed Ester-terminated glycidyl azide polymer[J].PolymerMaterialsScience&Engineering, 2012, 28(2): 1-4.
[11] 王晓, 罗运军, 柴春鹏. 端叠氮基聚叠氮缩水甘油醚的合成与热性能[J]. 火炸药学报,2010, 28(6): 57-60.
WANG Xiao, LUO Yun-jun, CHAI Chun-peng. Synthesis and thermal property of Azido-terminated glycidyl azide polymer[J].ChineseJournalofExplosives&Propellants, 2010, 28(6): 57-60.
[12] Pant C, Wagh R, Nair J, et al. Synthesis and characterization of first generation dendritic azidoesters[J].Propellants,Explosives,Pyrotechnics, 2007, 32(6): 461-467.
[13] Pant C S, Wagh R M, Nair J K, et al. Dendtritic azido ester: a potential energetic additive for high energy material (hem) formulations[J].JournalofEnergeticMaterials, 2006, 24(4): 333-339.
[14] 王晓, 柴春鹏, 罗运军. 端叠氮基超支化聚酯的合成与表征[J]. 高分子材料科学与工程, 2012, 28(1): 5-8.
WANG Xiao, CHAI Chun-peng, LUO Yun-jun. Synthesis and characterization of azido-terminated hyperbranched polyester[J].PolymerMaterialsScience&Engineering, 2012, 28(1): 5-8.
[16] Johannessen B. Low polydispersity glycidyl azide polymer: United States, 5741997[P]. 1998-04-21.
[17] Robert A E. Use of polymeric ethylene oxides in the preparation of glycidyl azide polymer: United States, 4486351[P]. 1984-12-04.
