Theoretical and Experimental Study of Nitrogen-rich Compounds of Biurea and 1-Amino-biurea
2015-05-10-,-,-,-
-, -, -, -
(State Key Laboratory of Explosion Science and Technology, Beijing Institute of Technology, Beijing 100081, China)
1 Introduction
Biurea is widely used in food additive and contamination analysis, researchers have conducted a study of this compound[1-3]. Biurea is also used industrially as a high-temperature blowing agent for expanding plastics such as polypropylene. The thermal decomposition of biurea is currently being studied and the crystal structure has been determined in order to provide evidence to explain the mechanism of the solid-state decomposition[4].
1-Amino-biurea is that we have just synthesized, with the amino substituted biurea end-group hydrogen atom[5]. Biurea and 1-amino-biurea both are thought to be useful materials and intermediates. As derivatives of hydrazine, they possess strong coordination capacity and reduction ability, and also play an important role as reducing agent in the metal complexes[6-7]. Since they are interesting azotic chain ligands with several lone-pair electron pairs that may coordinate with many metal ions and oxidizing groups as mono-dentate or multi-dentate ligands[8-12]. In addition, since this kind of derivatives may design a variety of energetic coordination compounds with explosive properties, they have gained more and more attentions as ligand with transition-metals especially in recent years in the areas of primary explosives, propellants, and high explosive[13-18].
This paper reports X-ray crystal of 1-amino-biurea, since the crystal structure of biurea has been determined[4]. To our knowledge, neither theoretical investigations nor comparison between the calculated and experimental results for the biurea and 1-amino-biurea compounds are available, which attract our attention and prompt us to make a study. Therefore, this paper reports the preparation and quantum chemical calculations of the two compounds. In addition, a comparison between the calculated results and experimental ones is performed, which may helpful for providing insight into the structures and properties of the title compounds and their derivatives.
2 Experimental and Computational Section
General caution:title compounds are energetic materials and tend to explode under certain conditions. Appropriate safety precautions should be taken, especially when these compounds are prepared on a larger scale.
2.1 Materials and Instruments
All the reagents used in the synthesis of title compounds were analytical grade and purchased commercially from Sinopharm Chemical Reagent Co., Ltd.
2.2 Synthesis of 1-amino-biurea
The urea (0.2 g, 20 mmol) was slowly added to a mixture of carbohydrazide(1.8 g, 20 mmol) and acetone (50 mL) at room temperature. After vigorous stirring in autoclave at 110 ℃ for 4 h, the resulting clear solvent was removed in vacuum. The product was recrystallized from water, in 65.3% yield. The slightly yellow single crystal with dimensions of 0.52 mm×0.28 mm×0.2 mm suitable for X-ray measurement were obtained by recrystallization of the products with distilled water at room temperature for 1 w.
2.3 X-ray crystallography
A Bruker Smart 1000 CCD diffractometer with graphite mono-chromated MoKαradiation (λ=0.071073 nm) was applied for structure analyses of the title compounds. The data were collected at 294(2) K usingφandωscan modes. A semi-empirical absorption correction was made using SADABS software[19]. The structure was solved using the direct methods and successive Fourier difference syntheses (SHELXS-97)[20], refined using full-matrix least-squares onF2with anisotropic thermal parameters for all non-hydrogen atoms (SHELXL-97)[21]. Hydrogen atoms were added theoretically and refined with riding model position parameters and fixed isotropic thermal parameters. Detailed information concerning crystallographic data collection and structure refinement is summarized in Table 1.
Table1Crystal data and structure refinement details of 1-amino-biurea

chemicalformulaC2H7N5O2formulamass133.13temperature/K294(2)crystalsystemOrthorhombicspacegroupP2(1)/nZ4a/Å9.194(1)b/Å4.756(1)c/Å12.665(2)V/Å3553.80(2)density(calculated)/g·cm-31.597absorptioncoefficient/mm-10.137F(000)/Å280θrangefordatacollection/(°)3.22to29.25h,k,andlrange0to12,0to6,-17to1reflectionmeasured1050independentreflection(Rint)854refinementmethodFull-matrixleast-squaresonF2data/restraints/parameters854/3/96goodness-of-fitonF20.897finalR1andwR2[I>2σ(I)]R1=0.0385,wR2=0.08491)R1andwR2indices(alldata)R1=0.0588,wR2=0.09191)largestdiff.peakandhole/e,Å-30.318,-0.211
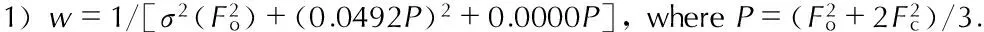
2.4 Computational methods
Based on crystal data, the structure optimization on the biurea and 1-amino-biurea compounds were carried out using the density functional theory (DFT) with the B3LYP method[22-23]employing the 6-31G**and cc-pVTZ basis sets[24-26]. In addition, the harmonic vibrational frequencies and infrared intensity were predicted at the B3LYP/cc-pVTZ level of theory DFT-B3LYP denoted the combination of the Becke′s three parameters hybrid functional with the Lee-Yang-Parr (LYP) correlation functional. The DFT method deals with the electron correlation but is still computationally economic. Because the B3LYP method was more widely used and tested, the hybrid density functional of B3LYP with the cc-pVTZ basis set was used for the calculations. The structure of 1-amino-biurea was fully optimized and the natural bond orbital analysis was performed on the optimized structure. The crystal structure of 1-amino-biurea obtained from the X-ray diffraction was used for the computation. All electronic structure calculations were performed with the Gaussian 03 program package.
3 Results and Discussion
3.1 The crystal structure of 1-amino-biurea
The crystal structure of 1-amino-biurea is shown in Fig.1. The shape of the 1-amino-biurea molecule can be explained by considering the following interactions. The repulsion of the lone pairs on the adjacent N atoms will be very strong due to theirp-πnature and a rotation about the N—N bond will reduce this interaction. However, as the N—N bond rotates the two C atoms start to approach each other. Thus final contact distance between these two C atoms was 3.380 Å(1-amino-biure), while 3.385 Å in biurea[4]. The further rotation about the N—C bonds may be imposed by the geometry of the hydrogen-bonding system.

Fig.1Molecular structure and atom label of 1-amino-biurea
Fig.2The packing of the molecule of 1-amino-biurea in crystal lattice
The obtained selected bond lengths and bond angles of 1-amino-biurea are summarized in Table 2. According to the bond lengths data of the compound, it can be concluded that the bond lengths of the N—N and C—N of the compounds are 1.386-1.409Å and 1.330-1.364Å, which are shorter than the general lengths (1.450 and 1.470Å). At the same time, the bond lengths of CO of 1-amino-biurea are the same 1.247Å, which are longer than the general length (1.230Å). So, the bond lengths of N—N, C—N, and CO tend to a homogeneous value, which is the result from thep-πconjugate effect between double bond of CO and thepelectronics of N atoms. From the data of bond angles, we can find that N(2)—N(1)—H(1A), N(2)—N(1)—H(1B) bond angles of 1-amino-biurea are close to 109°28′, so N(1) atom adoptssp3hybridized and H(1A), H(1B), N(2) form chemical bonds, and the othersp3hybrid orbital on a pair of lone pair electrons. Else N, C atoms of 1-amino-biurea are adoptedsp2hybridized, since the other bond angles are close to 120°.
Table2Selected bond lengths and bond angles of 1-amino-biurea
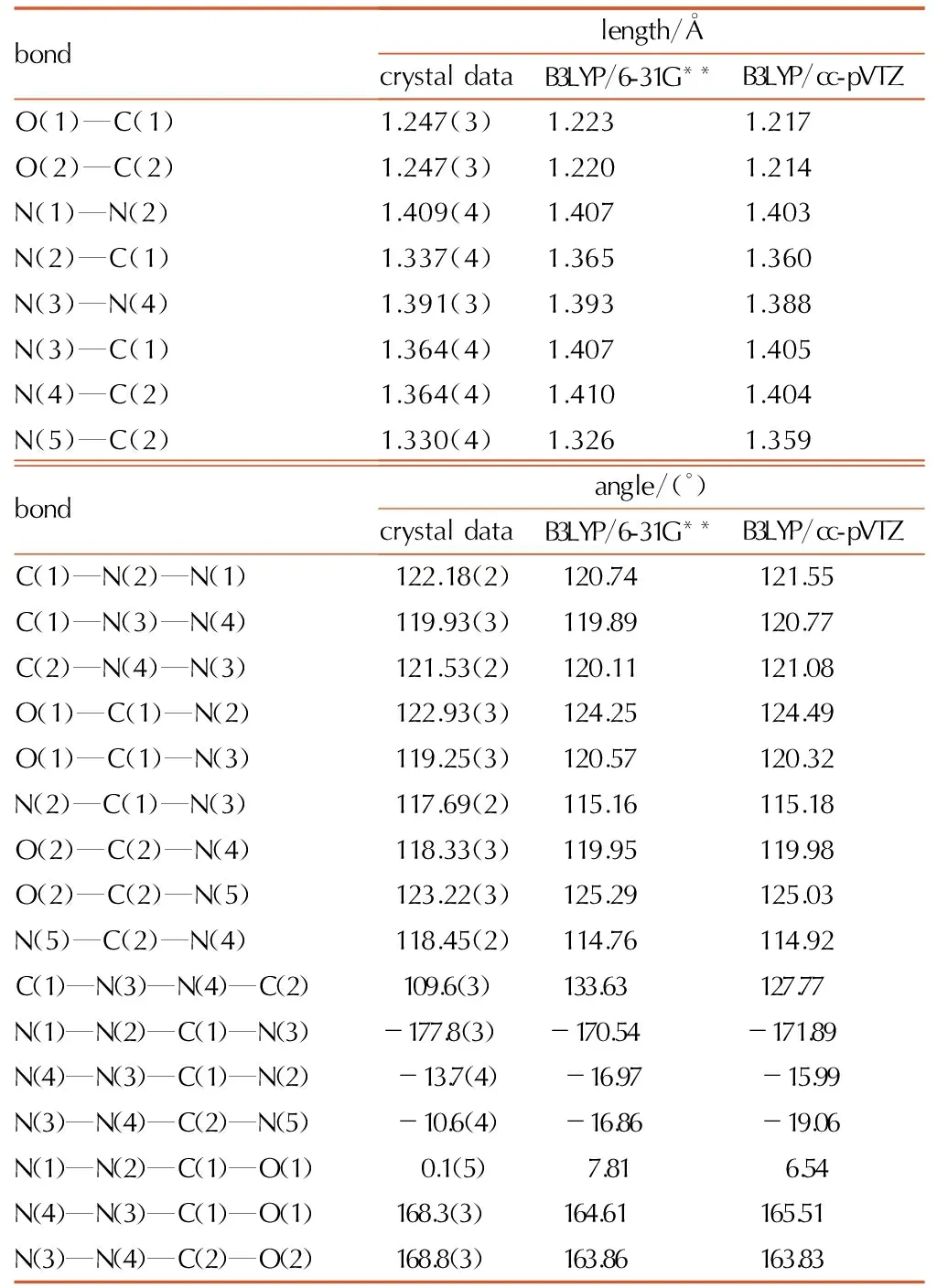
bondlength/ÅcrystaldataB3LYP/6-31G**B3LYP/cc-pVTZO(1)—C(1)1.247(3)1.2231.217O(2)—C(2)1.247(3)1.2201.214N(1)—N(2)1.409(4)1.4071.403N(2)—C(1)1.337(4)1.3651.360N(3)—N(4)1.391(3)1.3931.388N(3)—C(1)1.364(4)1.4071.405N(4)—C(2)1.364(4)1.4101.404N(5)—C(2)1.330(4)1.3261.359bondangle/(°)crystaldataB3LYP/6-31G**B3LYP/cc-pVTZC(1)—N(2)—N(1)122.18(2)120.74121.55C(1)—N(3)—N(4)119.93(3)119.89120.77C(2)—N(4)—N(3)121.53(2)120.11121.08O(1)—C(1)—N(2)122.93(3)124.25124.49O(1)—C(1)—N(3)119.25(3)120.57120.32N(2)—C(1)—N(3)117.69(2)115.16115.18O(2)—C(2)—N(4)118.33(3)119.95119.98O(2)—C(2)—N(5)123.22(3)125.29125.03N(5)—C(2)—N(4)118.45(2)114.76114.92C(1)—N(3)—N(4)—C(2)109.6(3)133.63127.77N(1)—N(2)—C(1)—N(3)-177.8(3)-170.54-171.89N(4)—N(3)—C(1)—N(2)-13.7(4)-16.97-15.99N(3)—N(4)—C(2)—N(5)-10.6(4)-16.86-19.06N(1)—N(2)—C(1)—O(1) 0.1(5) 7.81 6.54N(4)—N(3)—C(1)—O(1)168.3(3)164.61165.51N(3)—N(4)—C(2)—O(2)168.8(3)163.86163.83
Table3H-bond lengths and bond angles of 1-amino-biurea
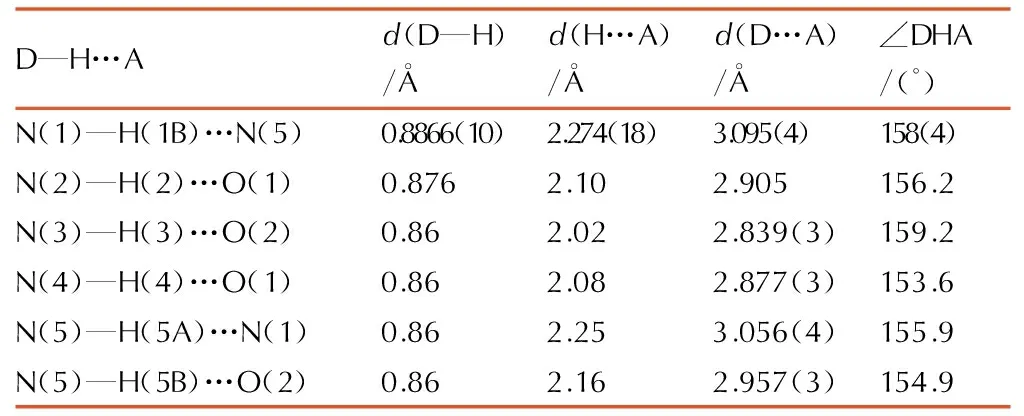
D—H…Ad(D—H)/Åd(H…A)/Åd(D…A)/Å∠DHA/(°)N(1)—H(1B)…N(5)0.8866(10)2.274(18)3.095(4)158(4)N(2)—H(2)…O(1)0.8762.102.905156.2N(3)—H(3)…O(2)0.862.022.839(3)159.2N(4)—H(4)…O(1)0.862.082.877(3)153.6N(5)—H(5A)…N(1)0.862.253.056(4)155.9N(5)—H(5B)…O(2)0.862.162.957(3)154.9
3.2 Quantum chemical calculation of biurea and 1-amino-biurea
The calculated data of 1-amino-biurea and biurea from B3LYP methods are shown in Table 2 and Table 4, respectively. The molecular structure and atom labels of biurea are shown in Fig.3. We can find the computational results obtained at B3LYP/6-31G**and B3LYP/cc-pVTZ level of theories are very similar. The B3LYP/ cc-pVTZ calculations give a remarkably good description of both of the molecular geometry, in which all bond distances of biurea deviate by less than 0.124 Å from experimental values, and the largest bond-angle error of biurea is 6.9°, while all bond distances of 1-amino-biurea deviate by less than 0.153 Å from experimental values, and the largest bond-angle error of 1-amino-biurea is 18.17°. These tiny differences are because that the gaseous molecule have been calculated in an ideal state of the most stable structure, and the calculation process does not take into account the interaction between molecules, while in the crystal structure exists in intermolecular hydrogen bonding, van der Waals effect.
The preferable sites for coordination in the title compounds are investigated from the theoretical results of Mulliken populations, NBO atomic charges and MESP under level of B3LYP/cc-pVTZ. These methods are proved to be accurate to predict to preferable coordination positions[27-29].
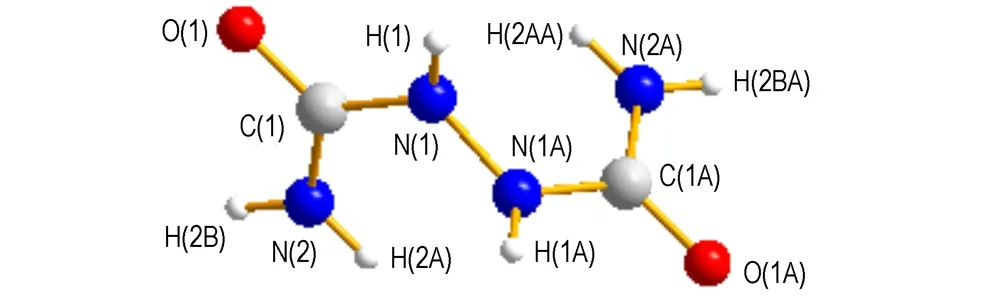
Fig.3The molecular structure and atom label of biurea
Table4Selected bond lengths and bond angles of biurea
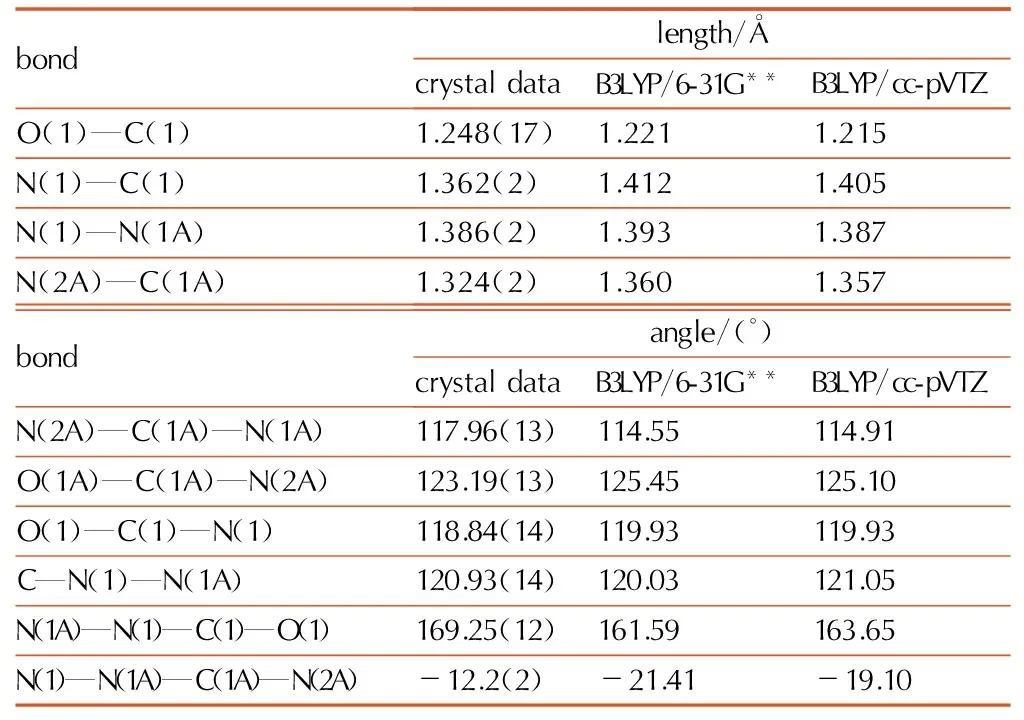
bondlength/ÅcrystaldataB3LYP/6-31G**B3LYP/cc-pVTZO(1)—C(1)1.248(17)1.2211.215N(1)—C(1)1.362(2)1.4121.405N(1)—N(1A)1.386(2)1.3931.387N(2A)—C(1A)1.324(2)1.3601.357bondangle/(°)crystaldataB3LYP/6-31G**B3LYP/cc-pVTZN(2A)—C(1A)—N(1A)117.96(13)114.55114.91O(1A)—C(1A)—N(2A)123.19(13)125.45125.10O(1)—C(1)—N(1)118.84(14)119.93119.93C—N(1)—N(1A)120.93(14)120.03121.05N(1A)—N(1)—C(1)—O(1)169.25(12)161.59163.65N(1)—N(1A)—C(1A)—N(2A)-12.2(2)-21.41-19.10
The harmonic vibrationalfrequencies and their infrared intensity of biurea and 1-amino-biurea are predicted at the B3LYP/ cc-pVTZ level of theory mentioned above, which all yield real frequencies for it. The predicted frequencies and intensities for biurea and 1-amino-biurea are listed in Table 5 and Table 6, respectively. All theoretical frequencies reported here are listed as calculated. Only the main vibrational frequencies of some functional groups have been assigned.
According toTable 5, the vibrational frequencies can be divided into three main absorption regions. Low frequency of less than 700 cm-1is the N—H bond of the rocking vibration absorption; medium frequency range of 900 cm-1to 1800 cm-1is N(2)—H bond of the swing plane symmetry vibration at approximately 900 cm-1, N(1)—H bond of the rocking vibration at about 1500 cm-1, N(2)—H bond of the shear vibration at about 1700 cm-1, as well as CO stretching vibration around 1800 cm-1; high frequency area of 3450 cm-1to 3650 cm-1is the N—H bond stretching vibration absorption area.
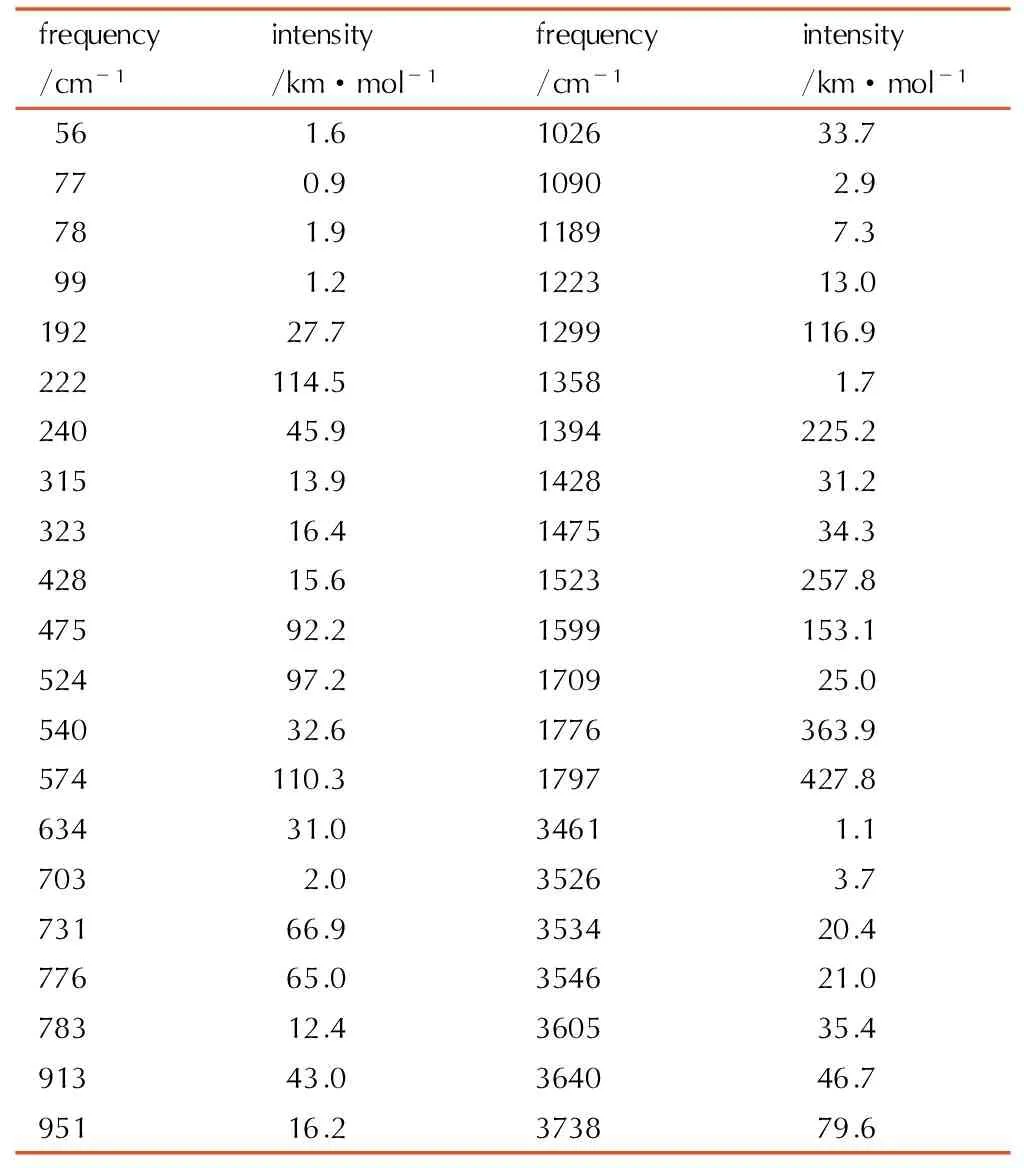
These characteristic absorption bandsshowed in Table 6 are shifted to lower wave number compared to the free ligand, and this indicates that the N atom of the hydrazine group and the carbonyl atom coordinate to the center cation. The important bands observed in the range of 1797 cm-1to 1358 cm-1and 783 cm-1to 634 cm-1are assigned to the symmetric stretching vibration and deformation vibration of the C—O bond. There are absorption peaks around 913 cm-1and 951 cm-1, which are assigned to the symmetric vibration of the C—N bond.
Table5A full vibrational assignment of biurea based on the B3LYP/ cc-pVTZ level of theory

frequency/cm-1Intensity/km·mol-1frequency/cm-1Intensity/km·mol-167 3.995123.581 3.31082 3.3100 0.11094 0.113440.21198 4.6235307.81385334.030215.31402108.1310 3.61438 6.3430 8.9148047.152054.61589246.5539 5.9160698.555070.41797803.3566 8.0179781.6577113.4354229.0651 6.9354413.074155.3360620.277364.9360856.177916.33742148.5950 0.1374211.8
Table6A full vibrational assignment of 1-amino-biurea based on the B3LYP/ cc-pVTZ level of theory
frequency/cm-1intensity/km·mol-1frequency/cm-1intensity/km·mol-156 1.6102633.777 0.91090 2.978 1.91189 7.399 1.2122313.019227.71299116.9222114.51358 1.724045.91394225.231513.9142831.232316.4147534.342815.61523257.847592.21599153.152497.2170925.054032.61776363.9574110.31797427.863431.03461 1.1703 2.03526 3.773166.9353420.477665.0354621.078312.4360535.491343.0364046.795116.2373879.6
Thecorrected factor of the vibrational frequencies calculated based on B3LYP/ cc-pVTZ level is 0.9614[30]. The corrected values of the calculated vibrational frequencies are basically in accordance with the experimental ones (biurea: 1570, 1660, 3160, 3430 cm-1)[31].
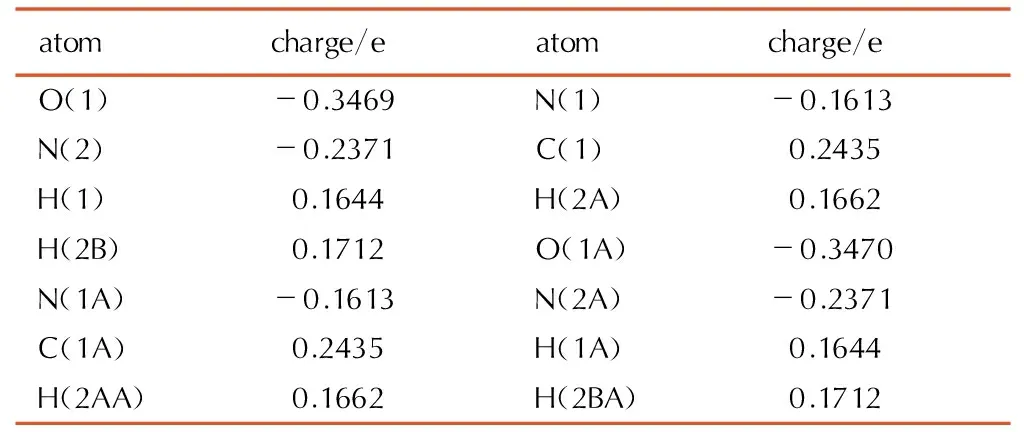
The selected Mulliken charge distribution data of biurea and 1-amino-biurea crystal are listed in Tables 7 and 8, respectively. Charge distribution of the carbon and hydrogen atoms are positively charged, nitrogen and oxygen atoms with a negative charge. This is mainly due to nitrogen and oxygen atoms of electro-negativity are relatively large, more capable to attract electrons. Since every N and O atom of biurea and 1-amino-biurea have a lone electron pair, all of them may be a potential coordination site. In biurea molecule, O atoms of the CO(N)-group have the most Mulliken charge of -0.3470e, followed by N(2A) atoms of the NH2(C)-group (-0.2371e) and the next is N(2) atom (-0.2371e). While, O(1) atom (-0.3700e) has the most Mulliken charge, secondly O(2) atoms(-0.3449e) of the CO(N) group and thirdly N(5) atoms (-0.2399e) of the NH2(C) group of 1-amino-biurea. Herein, O, N(2A) and N(2) atoms of biurea and O(1), O(2) and N(5) atoms of 1-amino-biurea are the most probable coordination sites. Expressed as bi-dentate ligand, which is nitrogen atom and carbonyl oxygen atom also participates in coordination.
Table7Selected Mulliken charge of biurea crystal at the B3LYP/ cc-pVTZ level
atomcharge/eatomcharge/eO(1)-0.3469N(1)-0.1613N(2)-0.2371C(1)0.2435H(1)0.1644H(2A)0.1662H(2B)0.1712O(1A)-0.3470N(1A)-0.1613N(2A)-0.2371C(1A)0.2435H(1A)0.1644H(2AA)0.1662H(2BA)0.1712
Table8Selected Mulliken charge of 1-amino-biurea crystal at the B3LYP/ cc-pVTZ level

atomcharge/eatomcharge/eO(1)-0.3700O(2)-0.3449N(1)-0.2382N(2)-0.1213N(3)-0.1665N(4)-0.1599N(5)-0.2399C(1)0.2557C(2)0.2434H(1A)0.1508H(1B)0.1557H(2)0.1723H(3)0.1619H(4)0.1650H(5A)0.1715H(5B)0.1645
The structures optimized by natural bond orbital (NBO) analysis, its atomic charge distribution of biurea in Table 9 and 1-amino-biurea in Table 10 are obtained. NBO atomic theory of orthogonal approach to determine the asymmetry between the atomic orbitals[22, 32], as compared with the Mulliken charge that it is given nothing to do with the basis set of basic NBO charge. Charge distribution was the same as the above-mentioned compounds, namely carbon, hydrogen atoms are positively charged, nitrogen and oxygen atoms with a negative charge. According to Table 9, all the N atoms in biurea molecule, NH2(C) based on the N (2) and N(2A) atom have the most NBO charge (-0.8159e), followed by O atoms of the CO(N)-group of biurea(-0.6339e). From Table 10, we can see that of all the atoms of 1-amino-biurea, N(5) atoms of the NH2(C)-group has the most NBO charge (-0.8172e), secondly O(1) atom (-0.6449e) and thirdly O(2) atoms(-0.6314e) of the CO(N)-group of 1-amino-biurea. O(1), O(2) and N(5) atoms of 1-amino-biurea are the most probable coordination sites. Therefore, NBO charge conformed the biurea ligand with metal ions of coordination is O, N(2), N(2A) atoms while, O(1), O(2) and N(5) atoms of 1-amino-biurea are the most probable coordination sites.
Table9Selected NBO charge of biurea crystal at the B3LYP/cc-pVTZ level

atomcharge/eatomcharge/eO(1)-0.6339N(1)-0.4937N(2)-0.8159C(1)0.7646H(1)0.3801H(2A)0.3995H(2B)0.3992O(1A)-0.6339N(1A)-0.4937N(2A)-0.8159C(1A)0.7646H(1A)0.3801H(2AA)0.3995H(2BA)0.3992
Table10Selected NBO charge of 1-amino-biurea crystal at the B3LYP/cc-pVTZ level

atomcharge/eatomcharge/eO(1)-0.6449O(2)-0.6314N(1)-0.6183N(2)-0.4575N(3)-0.4889N(4)-0.4917N(5)-0.8172C(1)0.7421C(2)0.7644H(1A)0.3466H(1B)0.3453H(2)0.3941H(3)0.3792H(4)0.3814H(5A)0.3991H(5B)0.3978
The molecular electrostatic potential (MESP) surface for biurea and 1-amino-biurea molecules calculated at B3LYP/cc-pVTZ level of theory, are given in Fig.4 and Fig.5, respectively. The red and blue color means positive and negative molecular electrostatic potential. It should be noted that the largest negative value of MESP does not necessarily correspond to the atom with the largest negative charge. In some cases, calculations of MESP allow to predict successfully the coordination sites in molecules[33]. From Fig.4 and Fig.5, it can be seen that, the nuclei naturally display the positive electrostatic potential (shown in red) on all molecules. The strong negative electrostatic potential (shown in blue) region associates with the lone pair of the carbonylgroup, which keep with the preceding NBO charges on atoms. In Fig.4, the highest negative values of the electrostatic potential are located near the N(2), N(2A) and O atoms of the carbonyl group of the biurea, and only a shallow minimum appears between the nitrogen atoms of the two amino groups. Again the negative potential occupies mainly on O(1), O(2) and N(5) atoms of the 1-amino-biurea, which may be attracted to the electrophiles. Therefore, the possible coordination sites in biurea molecule would be the O, N(2) and N(2A) atoms, while, O(1), O(2) and N(5) atoms of 1-amino-biurea are the most probable coordination site.
In order to study the possible coordination sites in biurea and 1-amino-biurea molecule under formation of complex compounds, three calculations have been carried out, what we can obtain that O, N(2) and N(2A) atoms of biurea are the most probable coordination sites, while O(1), O(2) and N(5) atoms of 1-amino-biurea are the most probable coordination site.
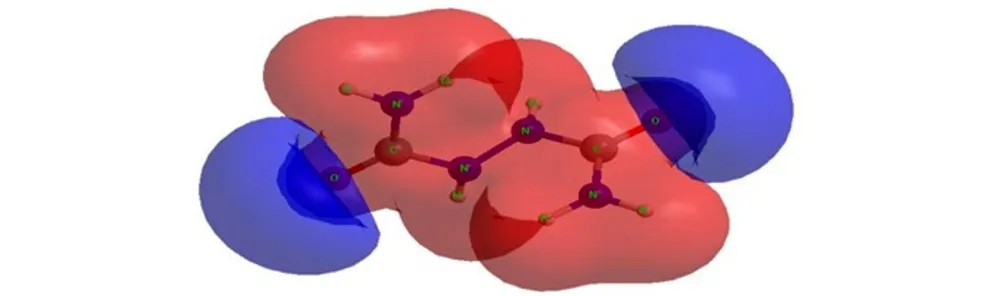
Fig.4MESP surface for biurea molecule calculated at B3LYP/cc-pVTZ level of theory
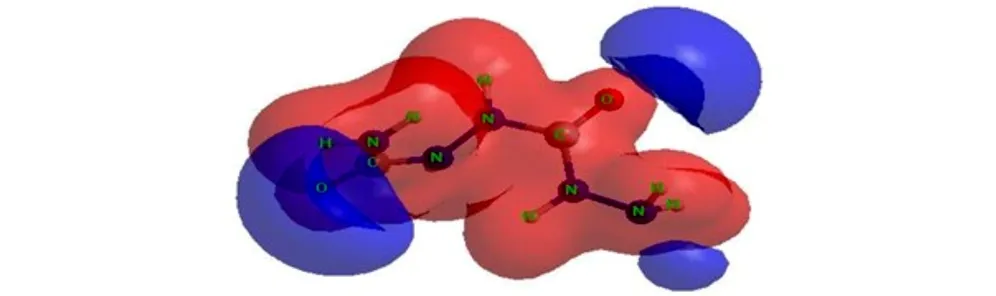
Fig.5MESP surface for 1-amino-biurea molecule calculated at B3LYP/cc-pVTZ level of theory
4 Conclusion
(1) The single crystal of 1-amino-biurea is cultured with slow evaporation method. The molecular structure and crystal structure of 1-amino-biurea are determined by X-ray single crystal diffraction analysis.
(2) DFT B3LYP method with cc-pVTZ basis set is employed to optimize the geometries of biurea and 1-amino-biurea compounds for the first time. The crystal structures of title compounds obtained from the X-ray diffraction are used for the computation with the Gaussian 03 program package. The computational results obtained at B3LYP/cc-pVTZ level of theories give a remarkably good description of the molecular geometry.
(3) Quantum-chemical calculations of Mulliken charge distribution and the NBO analysis result and molecular electrostatic potential for title compounds using B3LYP/ cc-pVTZ levels of theory show that the O, N(2) and N(2A) atoms of biurea are the most probable coordination site, while O(1), O(2) and N(5) atoms of 1-amino-biurea are preferable sites for metal coordination.
[1] Anklam E, Callede M B de la. Semicarbazide: occurrence in food products and state-of-the-art in analytical methods used for its determination[J].AnalBioanalChem, 2005, 382(4): 968-977.
[2] Pereira A S, Donato J L, Nucci G D. Implications of the use of semicarbazide as a metabolic target of nitrofurazone contamination in coated products[J]FoodAdditivesandContaminants, 2004(1), 21: 63-69.
[3] Mulder P P J, Beumera B, Van Rhijn J A. The determination of biurea: A novel method to discriminate between nitrofurazone and azodicarbonamide use in food products[J].AnalyticaChimicaActa,2007, 586(1-2) : 366-373.
[4] Brown B S, Russell P R. The crystal and molecular structure of biurea[J].ActaCryst, 1976, B32: 1056-1058.
[5] Gehlen L H, Dase G. Eine einfache synthese von 1-acyl-5-aminoformyl-carbohydraziden[J].EurJOrgChem, 1961, 646(1): 78-81.
[6] Wu B D, Li Y, Wang S W, et al. Preparation, crystal structure, thermal decomposition, and explosive properties of a novel energetic compound [Zn(N2H4)2(N3)2]n: a new high-nitrogen material (N=65.60%)[J].ZAnorgAllgChem, 2011, 637(3-4): 450-455.
[7] Qi S Y, Li Z M, Zhang T L, et al. Crystal Structure, thermal analysis and sensitivity property of [Zn(CHZ)3](ClO4)2[J].ActaChimSinica, 2011, 69(8): 987-992.
[8] Talawar M B, Agrawal A P, Chhabra J S, et al. Studies on lead-free initiators: synthesis, characterization and performance evaluation of transition metal complexes of carbohydrazide[J].JHazardMater, 2004(1-3), 113: 57-65.
[9] Wu B D, Zhang J G, Zhang T L, et al. Two environmentally friendly energetic compounds, [Mn(AZT)4(H2O)2](PA)2·4H2O and [Co(AZT)2(H2O)4](PA)2, based on 3-Azido-1,2,4-triazole (AZT) and picrate (PA) [J].EurJInorgChem, 2012, 8: 1261-1268.
[10] Sun Y H, Zhang T L, Zhang J G, et al. Kinetics of flash pyrolysis of [Co(CHZ)3](ClO4)2and [Ni(CHZ)3](ClO4)2[J].ActaPhysChimSin, 2006, 22(6): 649-652.
[11] Li Z M, Zhang G T, Zhang T L, et al. Synthesis, structural investigation and properties of a novel energetic coordination polymer [Pb(tza)2]n[J].ActaChimSinica, 2011, 69(10): 1253-1258.
[12] Liang Y H, Zhang J G, Cui Y, et al. Two novel nitrogen-rich energetic coordination compounds M2(DAT)5(H2O)3(TNR)2(M=Zn and Co): synthesis, characterization, thermal properties and sensitivity[J].ChinJStructChem, 2013, 31(3): 327-338.
[13] Zhang T L, Yang Y M, Zhang J G, et al. Preparation and molecular structure of [Pb2(TNR)(NO3)2(H2O)] [J].ChinJInorgChem, 2002, 18: 305-308
[14] Zhang J G, Li Z M, Zang Y, et al. Synthesis, structural investigation and thermal properties of a novel manganese complex Mn2(DAT)2Cl4(H2O)4(DAT=1,5-diaminotetrazole)[J].JHazardMater, 2010, 178(1-3): 1094-1099.
[15] Bushuyev O S, Brown P, Maiti A, et al. Ionic polymers as a new structural motif for high-energy-density materials[J].JAmChemSoc, 2012, 134(3): 1422-1425.
[16] Bushuyev O S, Peterson G R, Brown P, et al. Metal-organic frameworks (MOFs) as safer, structurally reinforced energetics[J].ChemEurJ, 2013, 19(5): 1706-1711.
[17] Wang S J, Tian Y W, You L X, et al. Synthesis, crystal structure and properties of a novel coordination polymer based on a trinuclear Mn(Ⅱ) cluster: [Mn3(bpta)2(bip)2]n[J].ChinJStructChem, 2013, 32(11): 1633-1638.
[18] Li Z M, Zhang T L, Zhang J G, et al. Synthesis, structure and thermal behaviors of a magnesium(Ⅱ) complex with tetrazole-1-acetic acid[J].ChinJStructChem, 2013, 32(7): 981-988.
[19] Sheldrick G M, ADABS, Version 2.03[CP]. University of Göttingen: Germany, 1996.
[20] Sheldrick G M, SHELXS-97, Program for the solution of crystal structure[CP]. University of Gottingen: Germany, 1997.
[21] Sheldrick G M, SHELXL-97, Program for the solution of crystal structure[CP]. University of Gottingen: Germany, 1997.
[22] Becke A D, Density-functional thermochemistry 3. The role of exact exchange[J].JChemPhys, 1993, 98: 5648-5652.
[23] Lee C, Yang W, Parr R G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density [J].PhysRevB, 1988, 37(2): 785-789.
[24] Dunning T H. Gaussian-basis sets for use in correlated molecular calculations .1. the atoms boron through neon and hydrogen[J].JChemPhys, 1989, 90(2): 1007-1023.
[25] Kendall R A, Dunning T J, Harrison R J. Electron-affinities of the 1st-row atoms revisited-systematic basis-sets and wave-fuctions[J].JChemPhy, 1992, 96: 6796-6806.
[26] Woon D E, Dunning T J. Gaussian-basis sets for use in correlated molecular calculations 3. the atoms aluminum through argon [J].JChemPhys, 1993, 98(2): 1358-1371.
[27] Zhang J G, Zhang T L, Yu K B. The preparation, molecular structure, and theoretical study of carbohydrazide (CHZ)[J].StructChem, 2006, 17(3): 249-254.
[28] Zhang J G, Zhang T L, Ma G X, et al. The crystal and computed structures of 1,2,4-triazol-5-one (TO)[J].JHeterocyclicChem, 2006, 43(2): 53-508.
[29] Xu C X, Yin X, Jin X, et al. Two coordination polymers with 3-hydrazino-4-amino-1,2,4-triazole as ligand: synthesis, crystal structures, and non-isothermal kinetic analysis[J].JCoordChem, 2014, 67(11): 2004-2015.
[30] Anthony P S, Leo R. Harmonic vibrational frequencies: an evaluation of hartree-fock, møller-plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors[J].JPhysChem, 1996, 100 (41): 16502-16513.
[31] Kirilin A D, Belova L O, Knyazev S P, et al. Organosilyl isocyanates. reactions with hydrazine, 1,1-dimethylhydrazine, and 1-methyl-1-[2-(1-methylhydrazino)-ethyl]-hydrazine and structural and electronic characteristics[J].RussJGeneChem, 2005, 75(12) : 1930-1934.
[32] Reed A E, Weinstock R B, Weinhold F. Natural- population analysis[J].JChemPhys,1985, 83: 735-746.
[33] Alcami M, Mo O, Yanez M. Enhanced Al+binding-energies of Some azoles-a theoretical-study of azole-X+(X=Na, K, Al) Complexes[J].JPhysChem, 1992, 96(7): 3022-3029.
