染色体22q11区域的基因组病
2015-05-04赵欣荣WeiminBiCheungSau
赵欣荣 Weimin Bi Cheung Sau W
·综述·
染色体22q11区域的基因组病
赵欣荣1Weimin Bi2Cheung Sau W2
基因组病是指由于人类基因组DNA的异常重排而引起临床表型的一类疾病[1]。人类的22号染色体是一个端着丝粒染色体,多个基因组病发生于22q11染色体区域。该区域富含具有高度同源性,导致基因组不稳定的低拷贝重复序列(low-copy repeats, LCRs),又称为片段重复(segmental duplications)。其中的8个LCRs是学者们关注的热点,也是研究较为详细的序列。由此,形成了2套命名体系,LCR22-A至LCR22-H或LCR22-2至LCR22-8等片段序列(图1)[2,3]。在基因组不同位置出现的LCRs配对并发生序列的交换,即非等位同源重组(nonallelic homologous recombination, NAHR),导致位于LCRs之间的序列重排,包括缺失、重复和倒位[4,5]。LCRs导致22q11区域不稳定,容易发生重排。大量22q11区域的不同大小的再发性重组已被报道,包括常见的3 Mb 22q11.2缺失、重复及芯片检测到的微小重排。位于22q11区域的致病基因由于重排导致剂量不平衡,引起临床表型,即基因组病。不断更新的文献报道详细地阐述了多个22q11区域基因组病的分子机制和临床表型。
本文主要总结近年来关于22q11区域基因组病遗传基础的研究报道,并着重阐述引起这些疾病的遗传来源,为临床能够发现和诊断该系列综合征提供信息。
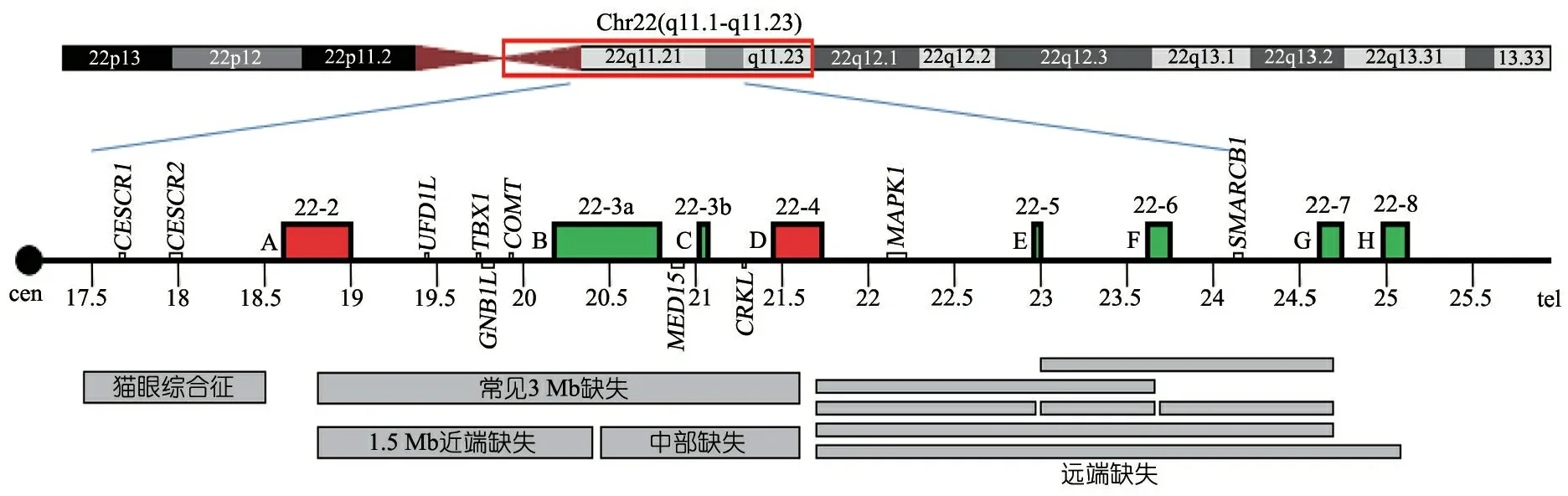
图1 22q11区域常见LCRs序列位点、缺失类型及关键基因位点(hg19)
1 22q11.2缺失综合征
22q11.2缺失综合征是最常见的微缺失综合征之一,新生儿的发生率约为1/4 000[6]。许多研究表明只有6%~25%的缺失是遗传的,大多数的缺失是新发的,提示22q11区域的突变率(~0.025%)极高[7~10]。22q11.2近端区域富含LCRs, LCR介导的NAHR导致了大多数22号染色体基因组异常[11]。这一系列综合征包括: 腭-心-面综合征(VCFS,OMIM 192430)、DiGeorge综合征(DGS,OMIM 188400),Cayler心面综合征(OMIM 125520)、圆锥动脉干异常脸综合征或Takao综合征(OMIM 217095),现统称为22q11.2缺失综合征[6]。其临床表型多样,包括心血管系统、腭部、泌尿系统和颅面部异常,发育、学习、智力、心理和行为障碍,免疫缺陷和先天性低血钙等[7,12,13]。
最常见的是3 Mb片段(LCR22-A至LCR22-D)缺失引起的综合征(图2)约占90%[14];其次是由LCR22-A和LCR22-B重组引起的1.5 Mb大小的22q11.2近端缺失,约占7%[14],由此产生的临床表型与3 Mb缺失引起的表型相近[15,16](图1);其他一些特异缺失或易位引起的缺失约占3%[14]。此外,位于10号染色体短臂10p13或附近的缺失也会引起类似DGS或VCFS的表型,称为DGS Ⅱ型[17],以区别于22q11.2缺失引起的DGS Ⅰ型。 DGS Ⅱ型的发生率极低,仅为DGS Ⅰ型的1/50[18]。
TBX1基因(OMIM 602504)单拷贝缺失是引起3 Mb和1.5 Mb这2种缺失表型的重要因素[12,13]。TBX1属于进化过程中保守的转录因子T-box家族成员之一,胚胎发生过程中其表达受到精确的调节。TBX1在器官发生时调节许多祖先细胞的分化和增生[19]。在基因敲除鼠模型中,TBX1缺乏导致心血管异常,咽弓动脉发育和成形的异常,心脏外流出道发育和分离异常,室间隔缺损和圆锥动脉干不分离[20]。TBX1调节口腔上皮细胞的黏附和腭部的发育[21],TBX1基因型的改变与DGS人群的腭裂表型相关[22]。TBX1重复或缺失的突变都与DGS相关[23]。

图2 常见22q11.2区域的LCRs介导的非等位同源重组模式图
Voss等[24]研究提示染色质修饰物-单核细胞白血病锌脂蛋白(MOZ/MYST3/KAT6A)是体内TBX1基因表达的重要调节者。MOZ是组蛋白乙酰基转移酶MYST家族的成员之一,在腭部、面部结构、胸腺和心血管系统发育中起重要作用。MOZ通过MOZ复合蛋白ING5与TBX1位点结合,影响其H3K9乙酰化,从而调节其表达。MOZ对于TBX1 mRNA正常转录是必需的,MOZ基因缺失导致复制叉转录因子无法维持正常水平,从而影响TBX1 mRNA正常转录。MOZ纯合突变或杂合突变同时合并TBX1单倍缺失都会导致类似DGS的症状,包括B型主动脉弓离断、胸腺发育异常、腭裂和室间隔缺损等。
22q11.2缺失综合征的成人中精神分裂症的发生率约为25%,而普通人群中1%的精神分裂症患者存在22q11.2缺失[25,26]。Van Beveren等[27]发现GNB1L、COMT、UFD1L和MED15等基因在22q11.2缺失综合征的患者中表达下降。COMT(OMIM 116790)编码儿茶酚氧位甲基移位酶,该酶在多巴胺代谢中起重要作用。COMT是精神分裂症易感性的强候选基因(OMIM 181500)[27,28]。GNB1L(OMIM 610778)编码G蛋白β亚单位样多肽,有报道证明GNB1L与精神分裂症相关[27,29]。GNB1L也与孤独症谱系障碍[30]和双向情感障碍有关[29]。UFD1L(OMIM 601754)编码泛素降解1样蛋白,泛素降解1样蛋白作用于泛素融合蛋白的降解。UFD1L的多态性也与精神分裂症相关[27]。此外,MED15多态性也与精神分裂症相关[27]。其他与22q11.2 DGS相关的精神分裂症的可能候选基因包括ZDHHC8(OMIM 608784)、PRODH(OMIM 606810)、RTN4R(OMIM 605566)、DGCR6(OMIM 601279)、DGCR6L(OMIM 609459)和ARVCF(OMIM 602269)等[31]。
2 22q11.2中部缺失综合征
LCR22-B与LCR22-D(距离1.5 Mb近端缺失相对较远,距离远端缺失相对较近)之间缺失的相关文献报道较少,通常称之为远端或非典型缺失[32~34]。为了避免与22q11.2远端缺失混淆,有学者建议称之为22q11.2中部缺失[6](图1)。鼠实验提示位于22q11.2中部的CRKL基因(OMIM 602007)缺失可引起相应22q11.2中部缺失综合征表型[35,36]。
Rump等[6]总结了52例22q11.2中部缺失综合征,其中有来自15个家族的27个病例为作者收集,另外25例为既往的文献报道。22q11.2中部缺失综合征心脏异常的发生率较低。腭裂和腭咽部缺陷、低血钙、低甲状旁腺激素等不是中部22q11.2缺失综合征患者的常见表型;小头畸形和生长受限发生率较高。智力障碍、行为问题、面部畸形和泌尿道异常等表型,在22q11.2中部缺失综合征和22q11.2缺失综合征中发生频率相近,提示22q11.2中部缺失综合征临床表型差异较大,很多症状与3 Mb缺失综合征共享,导致临床诊断困难。
大多数22q11.2中部缺失来源于LCR22-B与LCR22-C或LCR22-B与LCR22-D之间的重组。既往报道有3个家族的缺失超过了LCR22-D区域,与22q11.2远端缺失有重叠[33,34,37]。这些远端缺失的断裂点与已知的LCR片段不相关,提示其他的高度同源的片段作为替代底物参与重组[34]。这些病例的表型区别于其他病例,与22q11.2远端缺失综合征有更多相同的临床表型,也会出现早熟、产前或产后生长受限和小头畸形等表型[38,39]。
Rump等[12]还发现22q11.2中部缺失综合征的遗传率为57%,与之相比,22q11.2常见3 Mb片段缺失综合征的新发突变为90%~94%。同样,既往报道提示家族性遗传病例中1.5 Mb近端缺失出现频率也较高[40,41]。推测小的22q11.2近端缺失病例可能较大的3 Mb缺失病例有更好的生育力,小片段缺失较大片段缺失更有耐受力。由于两者之间在很多临床表型上有交叉,可采取统一的临床处理措施[42]。基于22q11.2中部缺失综合征遗传率较高,建议先证者的父母行缺失检测和遗传咨询。
3 22q11.2远端微缺失综合征
22q11.2远端微缺失综合征主要发生在3 Mb常见缺失区域远端的LCR22-D至LCR22-H之间,是由于断裂点位置不同而形成各种类型的可重复、独特的缺失综合征[38,39,43](图2)。主要临床表型为早熟、产前或产后生长受限、学习障碍、发育迟缓、特殊面容、中度骨骼异常和动脉干发育异常率增加等。与常见22q11.2缺失综合征相比较,22q11.2远端微缺失综合征病例的心脏缺陷发生率也较高,最常见的为室间隔缺损、各种瓣膜异位和动脉干发育异常。常见的外观异常包括扁平人中、耳结构异常、腭裂或双叉悬雍垂、小(缩)颌、睑裂斜上向外、薄上唇及耳前小赘肉等[39]。
位于LCR22-D与LCR22-E间的MAPK1/ERK2(OMIM 176948)基因与22q11.2远端微缺失综合征表型的相关性较大[39]。MAPK1/3(ERK1/2)是信号传导分子,通过细胞外刺激物,推动细胞膜至细胞浆和细胞核的运输[44]。MAPK1和MAPK3高度同源,共同受丝裂原活化蛋白激酶激活。MAPKs参与一系列生物事件,包括增生、分化、代谢、运动、生存和凋亡[45]。很多22q11远端缺失的病例缺失基因中包含MAPK1基因。MAPK1基因敲除小鼠会出现胎盘发育异常,从而导致宫内生长受限和宫内死亡[46]。
4 22q11.2重复综合征
LCRs错误配对后形成的不等交换,其结果是导致一条染色体的缺失和另一条染色体相应部位的重复(图2)。理论上重复和缺失的发生率应一致。尽管已经发现了22q11重复的病例,但与22q11缺失的发生率相比,明显少于预期。22q11重复的发生率仅为22q11缺失的50%[47]。大多数22q11.2微重复的病例携带约3 Mb与DGS/VCFS缺失区域相对应的重复,其他重复如1.5 Mb与DGS/VCFS近端缺失区域相对应的重复、部分4 Mb和6 Mb大的重复也见报道[48~50]。发育或语言障碍、行为问题、多发先天异常、轻度面容异常、听力缺失和生长迟缓等为22q11.2重复综合征的主要临床表型。这些个体的表型通常表现为轻中度异常且具高度异质性,有些病例表型基本正常或接近正常,有些病例会表现多个缺陷[47]。即便在同一家系中,表型差异亦很大,子代可表现为严重的心血管发育异常,而同样存在22q11.2重复的父亲仅有轻度的认知障碍[51]。需要发现更多的病例来丰富与重复相关的表型谱[47]。轻或中度表型患者常常未能及时就诊或确诊,这也可能是22q11.2重复综合征发生率偏低的一个原因[49]。
有些22q11.2重复综合征病例的临床表型与DGS/VCFS类似,包括心脏缺陷、泌尿系统异常、咽腭部缺陷伴或不伴腭裂等。动物实验中TBX1的高表达和低表达均会引起DGS/VCFS 表型。而且,TBX1突变导致的表达增加与TBX1重复的效果一致,在非缺失却在DGS/VCFS表型的病例中出现[52,53],或许可以解释为何22q11.2微缺失和微重复的病例有许多相似的表型[49]。
由于外显率及表达性的差异,22q11.2微重复病例表型的高度不一致性[49]。重复片段的大小与表型的严重程度是否相关尚不明确。动物实验研究提示,22q11.2区域基因的上下游调节元件之间的相互作用、基因组其他位置的基因通过基因调节途径等都会影响TBX1的表达[54]。除了基因表达水平的改变,其他机制如基因-环境相互作用、基因组印记等因素都可能在22q11.2微重复病例的表型中起重要作用。
5 猫眼综合征(CES)
CES于1965年由Schachenmann等学者命名[55], 发生率为1/50 000~1/150 000[56]。CES临床表型多样化,最常见的临床表型是耳前小赘物(81%~87%),肛门直肠异常(闭锁,前置,73%~81%),泌尿系统异常(如单侧肾脏缺失,71%),眼部缺损 (55%~61%)和先天性心脏异常(如肺静脉反流,法洛四联征,持续性左上腔静脉,瓣膜异常等,50%~63%)[57],智力发育通常正常或接近正常。41%的病例会出现3个主要的异常(眼部缺损、肛门异常和耳前发育异常)[58,59]。
由于小的额外标记染色体(small supernumerary marker chromosome,sSMC) 导致22号染色体长臂近端部分三体或四体是形成CES的主要原因[60],少数成因为CES关键区域(CESCR)的复制[61]。从着丝粒到 D22S57的CESCR区域大小约为2 Mb,其中位于17 659 680~17 702 744 Mb(hg19)之间的CESCR1和以17 956 630~18 033 845 Mb(hg19)为侧翼的CESCR2,分别为心/面、神经/眼部症状的主要候选基因[62](图1)。根据低拷贝复制(LCR22s)机制,可将CES分为Ⅲ型:Ⅰ型CES不涉及DGS关键区域,其缺失或重复的断裂点均位于22q11近端区域;Ⅱ型CES可涉及断裂点远端区域1个(非对称型,Ⅱa)或2个(对称型,Ⅱb),涵盖1个或2个拷贝的DGS关键区域[63];Ⅲ型CES的断裂点位于LCR22s外侧的远端,为双随体的sSMC[64]。
6 总结
综上所述,22号染色体q11区域的基因组病复杂多变,其表型谱极其广泛。其中最常见的VCFS,由于其临床表现典型,如精神分裂症、学习障碍等而受到学者们的关注。尽管在研究22q11基因组病表型及其发生机制方面已取得了很大的进步,但将研究结果转化至临床的应用还很少。
目前用于缺失或重复综合征诊断的技术主要有细胞核型分析、多重连接探针扩增技术、荧光原位杂交)、微阵列比较基因组杂交及新一代测序等。大多数实验室以aCGH为主要检测方法,其他方法可用于验证。CES(OMIM 115470)主效基因为CESCR1,CESCR2等,aCGH确切检出率未知,可通过aCGH检出多出的标志染色体;DiGeorge/VCFS/22q11.21缺失综合征(OMIM 188400)主效基因为TBX1等,aCGH检出率>95%;22q11.21重复综合征(OMIM 608363)主效基因为TBX1等,aCGH检出率约99%;22q11.2远端微缺失综合征(OMIM 611867)主效基因为MAPK1(ERK2),SMARCB1(INI1)等,aCGH检出率约99%[65]。相信随着科学的进步,检测方法和治疗方法的改善,能够帮助临床尽早发现病例,做到早发现、早诊断和早治疗。
[1]Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet,1998,14(10):417-422
[2]Shaikh TH, Kurahashi H, Saitta SC,et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet, 2000 ,9(4):489-501
[3]Dunham I, Shimizu N, Roe, BA, et al. The DNA sequence of human chromosome 22. Nature, 1999,402(6761):489-495
[4]Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet, 2006,7(2):85-97
[5]Shaffer LG, Lupski JR. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet ,2000,34:297-329
[6]Rump P, Leeuw ND, van Essen AJ, et al. Central 22q11.2 deletion. Am J Med Genet A, 2014 ,164(11):2707-2723
[7]Ryan AK, Goodship JA, Wilson DI, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet,1997,34(10):798-804
[8]Leana-Cox J, Pangkanon S, Eanet KR, et al. Familial DiGeorge/velocardiofacial syndrome with deletions of chromosome area 22q11: report of five families with a review of the literature. Am J Med Genet, 1996, 65(4):309-316
[9]McDonald-McGinn DM, LaRossa D, Goldmuntz E, et al. The 22q11.2 deletion: screening for deletion, diagnostic workup, and outcome of results. Report on 181 patients. Genet Test, 1997,1(2):99-108
[10]Emanuel BS. Molecular mechanisms and diagnosis of chromosome 22Q11.2 rearrangements. Dev Disabil Res Rev, 2008, 14(1): 11-18
[11]Shaikh TH, O'Connor RJ, Pierpont ME, et al. Low copy repeats mediate distal chromosome 22q11.2 deletions:sequence analysis predicts breakpoint mechanisms.Genome Res, 2007,17(4):482-491
[12]Lindsay EA. Chromosomal microdeletions: dissecting del22q11 syndrome. Nat Rev Genet, 2001 ,2(11):858-868
[13]McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine (Baltimore), 2011 ,90(1):1-18
[14]Edelmann L, Pandita RK, Spiteri E, et al. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum Mol Genet, 1999,8(7):1157-1167
[15]Carlson C, Sirotkin H, Pandita R, et al. Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am J Hum Genet,1997,61(3):620-629
[16]Bartsch O, Nemecková M, Kocárek E, et al. DiGeorge/velocardiofacial syndrome: FISH studies of chromosomes 22q11 and 10p14, and clinical reports on the proximal 22q11 deletion. Am J Med Genet A, 2003 , 117A(1):1-5
[17]Schuffenhauer S, Lichtner P, Peykar-Derakhshandeh P, et al. Deletion mapping on chromosome 10p and definition of a critical region for the second DiGeorge syndrome locus (DGSII). Eur J Hum Genet, 1998, 6(3):213-225
[18]Berend SA, Spikes AS, Kashork CD, et al. Dual-probe fluorescence in situ hybridization assay for detecting deletions associated with VCFS/DiGeorge syndrome Ⅰ and DiGeorge syndrome Ⅱ loci. Am J Med Genet, 2000, 91(4):313-317
[19]Gao S, Li X, Amendt BA. Understanding the Role of Tbx1 as a Candidate Gene for 22q11.2 Deletion Syndrome. Curr Allergy Asthma Rep , 2013, 13(6):613-621
[20]Vitelli F, Morishima M, Taddei I, et al. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet, 2002, 11(8):915-922
[21]Funato N, Nakamura M, Richardson JA, et al. Tbx1 regulates oral epithelial adhesion and palatal development. Hum Mol Genet, 2012, 21(11):2524-2537
[22]Herman SB, Guo T, McGinn DM, et al. Overt cleft palate phenotype and TBX1 genotype correlations in velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome patients. Am J Med Genet A, 2012, 158A(11):2781-2787
[23]Yagi H, Furutani Y, Hamada H, et al. Role of TBX1 in human del 22q11.2 syndrome. Lancet, 2003, 362(9393):1366-1373
[24]Voss AK, Vanyai HK, Collin C, et al. MOZ regulates the Tbx1 locus, and Moz mutation partially phenocopies DiGeorge syndrome. Dev Cell, 2012, 23(3):652-63
[25]Bassett AS, Chow EWC, Husted J, et al. Clinical features of 78 adults with 22q11 Deletion Syndrome. Am J Med Genet A, 2005,138(4):307-313
[26]Bassett AS, Chow EW. Schizophrenia and 22q11.2 Deletion Syndrome. Curr Psychiatry Rep, 2008, 10(2): 148-157
[27]van Beveren NJ, Krab LC, Swagemakers S, et al. Functional gene-expression analysis shows involvement of schizophrenia-relevant pathways in patients with 22q11 deletion syndrome. PLoS One, 2012, 7(3):e33473
[28]Ira E, Zanoni M, Ruggeri M, et al. COMT, neuropsychological function and brain structure in schizophrenia: a systematic review and neurobiological interpretation. J Psychiatry Neurosci, 2013, 38(6):366-380
[29]Li Y, Zhao Q, Wang T, et al. Association study between GNB1L and three major mental disorders in Chinese Han populations.Psychiatry Res, 2011, 187(3):457-459
[30]Chen YZ, Matsushita M, Girirajan S, et al. Evidence for involvement of GNB1L in autism. Am J Med?Genet?B Neuropsychiatr? Genet, 2012, 159B(1):61-71
[31]Chen CP, Huang JP, Chen YY, et al. Chromosome 22q11.2 deletion syndrome: prenatal diagnosis, array comparative genomic hybridization characterization using uncultured amniocytes and literature review. Gene, 2013,527(1):405-409
[32]Fernández L, Nevado J, Santos F, et al. A deletion and a duplication in distal 22q11.2 deletion syndrome region. Clinical implications and review. BMC Med Genet, 2009, 10:48
[33]Garavelli L, Rosato S, Wischmeijer A, et al. 22q11.2 Distal deletion syndrome: description of a new case with truncus arteriosus type 2 and review. Mol Syndromol, 2011, 2(1):35-44
[34]Breckpot J, Thienpont B, Bauters M, et al.Congenital heart defects in a novel recurrent 22q11.2 deletion harboring the genes CRKL and MAPK1.Am J Med Genet A, 2012,158A(3):574-580
[35]Guris DL, Fantes J, Tara D, et al. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet, 2001,27(3):293-298
[36]Moon AM, Guris DL, Seo JH, et al. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell, 2006,10(1):71-80
[37]Ogilvie CM, Ahn JW, Mann K, et al. A novel deletion in proximal 22q associated with cardiac septal defects and microcephaly: a case report. Mol Cytogenet, 2009, 2:9
[38]Ben-Shachar S, Ou Z, Shaw CA, et al. 22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am J Hum Genet, 2008,82(1):214-221
[39]Fagerberg CR, Graakjaer J, Heinl UD, et al. Heart defects and other features of the 22q11 distal deletion syndrome. Eur J Med Genet, 2013,56(2):98-107
[40]Adeyinka A, Stockero KJ, Flynn HC, et al. Familial 22q11.2 deletions in DiGeorge/velocardiofacial syndrome are predominantly smaller than the commonly observed 3Mb. Genet Med, 2004,6(6):517-520
[41]Fernández L, Lapunzina P, Pajares IL, et al. Higher frequency of uncommon 1.5-2 Mb deletions found in familial cases of 22q11.2 deletion syndrome. Am J Med Genet A, 2005,136(1):71-75
[42]Bassett AS, McDonald-McGinn DM, Devriendt K, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr, 2011, 159(2):332-339.e1
[43]Rødningen OK, Prescott T, Eriksson AS, et al. 1.4Mb recurrent 22q11.2 distal deletion syndrome, two new cases expand the phenotype. Eur J Med Genet, 2008, 51(6):646-650
[44]Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev, 2001,22(2):153-183
[45]Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev, 2010,90(4):1507-1546
[46]Aouadi M, Binetruy B, Caron L, et al. Role of MAPKs in development and differentiation: Lessons from knockout mice. Biochimie, 2006,88(9):1091-1098
[47]Portnoï MF. Microduplication 22q11.2: A new chromosomal syndrome. Eur J Med Genet, 2009,52(2-3):88-93
[48]Alberti A, Romano C, Falco M, et al. 1.5 Mb de novo 22q11.21 microduplication in a patient with cognitive deficits and dysmorphic facial features. Clin Genet, 2007,71(2):177-182
[49]Ou Z, Berg JS, Yonath H, et al. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet Med, 2008,10(4):267-277
[50]Yu S, Cox K, Friend K, et al. Familial 22q11.2 duplication: a three-generation family with a 3-Mb duplication and a familial 1.5-Mb duplication. Clin Genet, 2008,73(2):160-164
[51]de La Rochebrochard C, Joly-Hélas G, Goldenberg A, et al. The intrafamilial variability of the 22q11.2 microduplication encompasses a spectrum from minor cognitive deficits to severe congenital anomalies. Am J Med Genet A , 2006,140(14): 1608-1613
[52]Torres-Juan L, Rosell J, Morla M, et al. Mutations in TBX1 genocopy the 22q11.2 deletion and duplication syndromes: a new susceptibility factor for mental retardation. Eur J Hum Genet, 2007,15(6):658-663
[53]Zweier C, Sticht H, Aydin-Yaylagül I, et al. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am J Hum Genet, 2007,80(3):510-517
[54]Aggarwal VS, Morrow BE. Genetic modifiers of the physical malformations in velo-cardio-facial syndrome/DiGeorge syndrome. Dev Disabil Res Rev, 2008,14(1): 19-25
[55]Schachenmann G, Schmid W, Fraccaro M, et al. Chromosomes in coloboma and anal atresia. Lancet ,1965,2(7406):290
[56]Kvarnung M, Lindstrand A, Malmgren H,et al. Inherited mosaicism for the supernumerary marker chromosome in cat eye syndrome: inter- and intra-individual variation and correlation to the phenotype. Am J Med Genet A, 2012,158A(5):1111-1117
[57]Belangero SI, Bellucco FT, Cernach MC, et al. Interrupted aortic arch type B in A patient with cat eye syndrome. Arq Bras Cardiol, 2009, 92(5): e29-31, e56-58
[58]Berends MJ, Tan-Sindhunata G, Leegte B, et al. Phenotypic variability of cat-eye syndrome. Genet Couns, 2001, 12(1): 23-34
[59]Rosias PR, Sijstermans JM, Theunissen PM, et al. Phenotypic variability of the cat eye syndrome. Case report and review of the literature. Genet Couns, 2001, 12(3): 273-282
[60]Jezela-Stanek A, Dobrzańska A, Maksym-Gasiorek D, et al. Trisomy 22pter-q12.3 presenting with hepatic dysfunction variability of cat-eye syndrome. Clin Dysmorphol, 2009,18(1): 13-17
[61]Meins M, Burfeind P, Motsch S, et al. Partial trisomy of chromosome 22 resulting from an interstitial duplication of 22q11.2 in a child with typical cat eye syndrome. J Med Genet, 2003, 40(5):e62
[62]Haltrichó I, Pikó H, Kiss E, et al. A de novo atypical ring sSMC(22) characterized by array CGH in a boy with cat-eye syndrome. Mol Cytogenet, 2014 ,7:37
[63]McTaggart KE, Budarf ML, Driscoll DA, et al. Cat eye syndrome chromosome breakpoint clustering: identification of two intervals also associated with 22q11 deletion syndrome breakpoints. Cytogenet Cell Genet, 1998,81(3-4):222-228
[64]Bartsch O, Rasi S, Hoffmann K, et al. FISH of supernumerary marker chromosomes (SMCs) identifies six diagnostically relevant intervals on chromosome 22q and a novel type of bisatellited SMC(22). Eur J Hum Genet, 2005,13(5):592-598
[65]Yu S, Graf WD, Shprintzen RJ. Genomic disorders on chromosome 22. Curr Opin Pediatr, 2012, 24(6):665-671
(本文编辑:张萍)
10.3969/j.issn.1673-5501.2015.05.013
1 上海交通大学医学院附属国际和平妇幼保健院 上海,200030;2 美国贝勒医学院分子和人类遗传学系 休斯顿,77030
Cheung Sau W,E-mail:scheung@bcm.edu
2015-01-15
2015-07-09)
