HPLC法测定依托度酸胶囊的含量
2015-03-15王增日
王增日
(鲁南制药集团股份有限公司,山东临沂276000)
HPLC法测定依托度酸胶囊的含量
王增日
(鲁南制药集团股份有限公司,山东临沂276000)
目的建立高效液相色谱法测定依托度酸胶囊中依托度酸含量的方法。方法采用高效液相色谱法,色谱柱为Diamonsil C18柱(4.6 mm×150 mm,5μm);乙腈:水+磷酸[520∶480(500+0.25)]为流动相,流速1.5 mL·min-1,检测波长为274 nm。结果在选定的色谱条件下依托度酸胶囊中辅料对主药无干扰,依托度酸在24.98~199.8μg·m L-1浓度范围内峰面积与浓度线性关系良好,相关系数r=0.999 9,平均回收率为99.6%,RSD为0.47%(n=9)。结论本方法简便、准确,专属性强,可用于依托度酸胶囊中依托度酸的含量测定。
依托度酸;高效液相色谱法;含量测定;胶囊
1985年,依托度酸由Wyeth Pharms Inc(惠氏制药公司)首次在英国上市,随后在美国等国上市,其上市剂型为片、胶囊(200~500 mg),缓释片(400~500 mg)[1]。依托度酸为非甾体抗炎药,具有抗炎、解热和镇痛作用。其作用机理可能是通过阻断环氧合酶的活性,从而抑制了前列腺素(PG)的合成。本品为非类固醇抗炎药(NSAIDs),作为镇痛及消炎药,其疗效可与阿司匹林、其他镇痛药及许多目前最常用的处方用NSAIDs相比[2]。文献有报道[3~7],用紫外分光光度法测定依托度酸片半成品的含量,依托度酸缓释片含量及其甲基衍生物的高效液相色谱(HPLC)法测定,HPLC法测定依托度酸双层片中主药的含量,HPLC法测定依托度酸及其在大鼠的药动学,HPLC法测定依托度酸缓释片中的有关物质,有关依托度酸血药浓度测定研究国外有报道[8~11],作者未见有HPLC法测定依托度酸胶囊的文献。本试验采用HPLC法测定依托度酸胶囊含量,方法简便、专属性强,结果准确,制剂中的辅料对分离无影响。
1 仪器、试药及试剂
Agilent1100高效液相色谱仪;VWD检测器;Mettler Toledo AG285电子天平;JCX-600GZ超声波清洗机;依托度酸对照品(中国药品生物制品检定所,批号:100667-200401,含量:100.0%);依托度酸胶囊200 mg样品(山东新时代药业有限公司,批号:012110601、012110602、012110603);乙腈为色谱纯,磷酸为分析纯,试验用水为二次蒸馏水。
2 方法与结果
2.1 溶液配制
2.1.1 色谱条件 色谱柱为Diamonsil C18柱(4.6mm×150 mm,5μm);乙腈:水+磷酸[520∶480(500+0.25)]为流动相,流速1.5 mL·min-1,检测波长为274 nm,进样量20μL。
2.1.2 溶液制备 对照品溶液的制备:精密称取依托度酸对照品25 mg,置100 mL量瓶中,加流动相适量,振摇使依托度酸溶解,加流动相稀释至刻度,摇匀,制成含依托度酸0.25 mg·mL-1的溶液,作为对照品储备溶液。精密量取储备液2 mL,置5 mL量瓶中,加流动相稀释至刻度,摇匀,即得含依托度酸0.10 mg·mL-1的对照品溶液。
供试品溶液的制备:取依托度酸胶囊内容物,精密称定,研细,精密称取细粉适量(约相当于依托度酸10 mg),置100 mL量瓶中,加流动相适量,振摇使依托度酸溶解,加流动相稀释至刻度,摇匀,滤过,作为供试品溶液。
空白溶液:按处方量取除主药成分的辅料,按供试品溶液制备方法制备,作为空白溶液。
2.2 系统适用性试验 精密量取“2.1.2”项下对照品溶液,理论塔板数不低于2 000,分离度均大于1.5。
2.3 空白试验 取供试品溶液、对照品溶液和空白溶液,按上述色谱条件进行测定,供试品溶液主峰的保留时间与对照品溶液主峰的保留时间一致,且空白辅料无干扰,见图1。
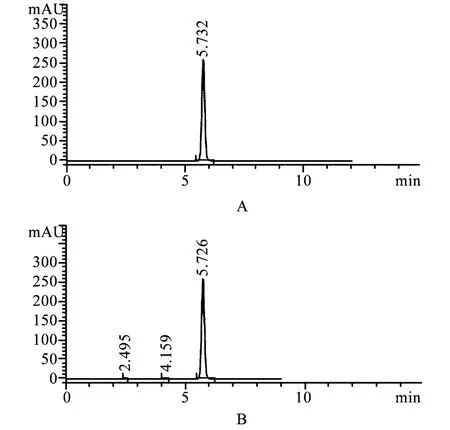
图1 HPLC色谱图A.对照品溶液;B.供试品溶液
2.4 线性关系 精密吸取对照品储备溶液0.5、1.0、2.0、3.0和4.0 mL,分别置5 mL量瓶中,再分别加流动相稀释至刻度,摇匀,分别进样。以依托度酸浓度(μg·mL-1)为横坐标,依托度酸峰面积为纵坐标,绘制标准曲线,计算回归方程为:Y=70.951X+143.66,r=0.999 9。其中,Y为依托度酸峰面积,X为依托度酸浓度(μg·mL-1)。结果指出在24.98~199.8μg·mL-1浓度范围内有良好的线性关系。
2.5 精密度试验 取同一对照品溶液,按上述试验条件,同日内测定6次,依托度酸的峰面积的RSD为0.43%。连续5 d(每天进样1次)进行测定,依托度酸的峰面积的RSD为0.77%。
2.6 稳定性试验 取同一供试品溶液,按上述试验条件于配制后0、2、4、6、8 h测定,依托度酸的峰面积的RSD为0.49%,表明供试品溶液在8 h内稳定。
2.7 重复性试验 对同一批号样品分别取样6份,按“2.1.2”项下方法配制6份供试品溶液,分别按上述试验条件测定,按外标法以依托度酸峰峰面积计算含量,含量结果的RSD为0.53%。
2.8 回收率试验 按处方量取高(120%)、中(100%)、低(80%)3种浓度的依托度酸对照品,每个浓度分别称3份均加入处方量辅料,制成供试样品,按“2.1.2”项下方法配制供试品溶液。分别精密量取供试品溶液与对照品溶液各20μL注入液相色谱仪,按上述色谱条件,测得回收率为99.6%,RSD=0.47%(n=9)。
2.9 样品含量测定 取本品3批,按“2.1.2”项下制备供试品溶液并按上述试验条件测定,计算依托度酸胶囊中依托度酸的含量。结果见表1。
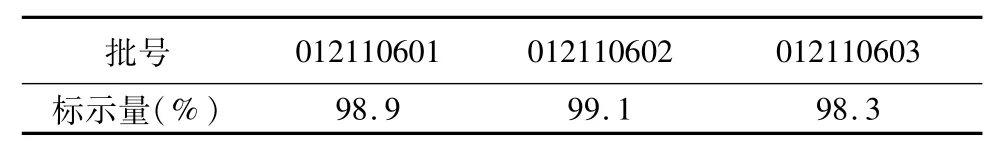
表1 样品结果测定结果
3 讨论
本文建立了HPLC法测定依托度酸胶囊含量的方法,依托度酸在225 nm、274 nm处有最大吸收,且在274 nm处辅料没有干扰,故检测波长定为274 nm,对测定没有影响,保证了方法的专属性。
[1] 徐梅桔,方晓萍,史磊.高效液相色谱法测定依托度酸的含量[J].广东化工,2013,40(5):19-20.
[2] 朱孙祥,余良水.高效液相色谱法测定依托度酸的含量[J].科技创新导报,2009,10:1.
[3] 李宋平.紫外分光光度法测定依托度酸片半成品的含量[J].广东药学院学报,2000,16(2):111-112.
[4] 黄志红,方原.依托度酸缓释片含量及其甲基衍生物的HPLC法测定[J].中国医药工业杂志,2001,32(4):167-169.
[5] 吴国民.HPLC法测定依托度酸双层片中主药的含量[J].现代医药卫生,2010,26(4):483-484.
[6] 石劲敏,许长江,段更利,等.高效液相色谱法测定依托度酸及其在大鼠的药动学[J].中国新药与临床杂志,2002,21(4):193-196.
[7] 杨婉花.高效液相色谱法测定依托度酸缓释片中的有关物质[J].中国药房,2005,16(17):1330-1332.
[8] Cosyns L,Spain M.Kraml M.Sensitive high-performance liquid chromatographic method for the determination of etodolac in serum[J].JPharm Sci,1983,72(3):275-277.
[9] Jamali F,Mehvar R.Lemko C,et al.Application of stereospecific high-performance liquid chromatography assay to a pharmacokineti study of etodolac enantiomers in humans[J].JPharm Sci,1988,77(11):963-966.
[10]Becker-Scharfenkamp U,Blaschke G.Evaluation of the stereoselective metabolism of the chiral analgesic drug etodolac by high-performance liquid chromatography[J].JChromatogr,1993,621(2):199-207.
[11]Molina-Martinez IT,Herrero R,Gutiérrez JA,et al.Bioavailability and bioequivalence of two formulations of etodolac(tablets and suppositories)[J].J Pharm Sci,1993,82(2):211-213.
Determ ination of etodolac in Etodolac Capsules by HPLC
WANG Zeng-ri
(Lunan Pharmaceutical Group Limited Company,Linyi276000,China)
ObjectiveTo establish an HPLCmethod for the determination of etodolac in Etodolac Capsules.MethodsHPLCmethod was adopted with Diamonsil C18column(4.6 mm×150 mm,5μm).A mixture of acetonitrile:water+phosphoric acid[520∶480(500+0.25)]was used as themobile phase.The flow rate was 1.5 mL·min-1with the detection wavelength at274 nm.ResultsAt the validatedmethod,the excipienthad no interference to the principalagent.A linear range of etodolac was 24.98~199.8μg·mL-1(r=0.999 9).The average recovery was 99.6%with RSD=0.47%(n=9).ConclusionThismethod was simple,accurate,specific and could be used for the determination of etodolac in Etodolac Capsules.
Etodolac;HPLC;Determination;Capsule
R927.2
A
2095-5375(2015)08-0451-003
王增日,男,工程师,研究方向:药物研发与注册,E-mail:1157014084@qq.com
