膜果麻黄种子不同发育时期的转录组测序分析1)
2015-03-08邓楠史胜青常二梅刘建锋兰倩江泽平
邓楠 史胜青 常二梅 刘建锋 兰倩 江泽平
(中国林业科学研究院林业研究所,北京,100091)
责任编辑:潘 华。
麻黄属(Ephedra)植物属于裸子植物门买麻藤目麻黄科,多为灌木、半罐木或草本植物[1],麻黄属全世界约有40 种,主要分布于亚洲、美洲、欧洲东南部及非洲北部等干旱荒漠地区。我国有12 种,4 变种[2],属于超旱生、强旱生、旱生系列植物,是西北荒漠区的主要建群种,在防风固沙、稳定植被方面起着非常重要的作用[3]。该属植物不仅与其同类的2种买麻藤类植物(买麻藤(Gnetum)和百岁兰(Welwitschia))在系统进化位置上特殊[4],还以富含麻黄碱而多为药用,尤其是草麻黄(E.sinica)和中麻黄(E.intermedin),最早公元前2700年就已用于治疗多种疾病[5],其中主要用于支气管炎和中枢神经系统的兴奋剂[6]。
然而,膜果麻黄(E.przewalskii)由于几乎不含有麻黄碱,传统上常作为植被恢复的优良抗逆材料。而在植物的整个生命周期中,种子的阶段最能够适应不良环境,是植物对于逆境适应程度最好的反映[7]。随着分子生物技术的发展,对于麻黄属植物的利用与研究迫切需要它的遗传信息。因此,在该属植物基因信息相对匮乏情况下开展其转录组研究就显得尤为重要。随着第二代高通量测序技术(NGS:Next Generation Sequencing technology)发展,基因研究的速度得到大幅提升[8-9]。这不仅保证了高质量测序数据,而且测序成本不到以前的1%[10]。由于转录组能显示在特定时期基因的表达状况,是基因挖掘以及研究生理响应相关各过程的有利工具[11],并且非常适合于基因组图谱尚未完成以及遗传数据信息匮乏的物种[12-14]。因此,本研究以不同发芽时期的膜果麻黄种子为研究对象,利用高通量RNA-seq 技术对其转录组进行测序分析,全面探讨不同发育时期中麻黄种子转录组基本信息,将有利于发掘麻黄种子萌发过程中抗逆相关基因及其种子中重要生物活性成分代谢关键基因,这对其优良性状的遗传改良及其植被恢复具有重要意义。
1 材料与方法
膜果麻黄的种子采集于甘肃省民勤县沙漠区(101°49'41″~104°12'10″E;38°3'45″~39°27'37″N),大陆性沙漠气候特征明显,平均海拔为1 400 m。对采集的种子选择大小均匀的个体置于培养皿中湿润的滤纸上进行发芽处理,处理时间为0、1.5、3、6、9、18 h,每个处理100 颗种子。
1.1 RNA 提取及cDNA 合成
不同萌发时期种子的总RNA 提取采用Trizol Reagent 方法(Invitrogen),并用DNaseΙ 进行DNA 消化处理,随后检测总RNA 完整性和质量(1.2%琼脂凝胶电泳);然后将各个发芽阶段的总RNA 进行等量混合后,进行mRNA 分离纯化(Dynabeads mRNA Purification Kit,Invitrogen)。用带有Oligo(dT)的磁珠富集真核生物mRNA。加入fragmentation buffer将mRNA 打断成短片段,以mRNA 为模板,用六碱基随机引物(random hexamers)合成第一条cDNA链,然后加入缓冲液、dNTPs、RNase H 和DNA polymerase I 合成第二条cDNA 链,在经过QiaQuick PCR试剂盒纯化并加EB 缓冲液洗脱之后做末端修复、加poly(A)并连接测序接头,然后用琼脂糖凝胶电泳进行片段大小选择,最后进行PCR 扩增,建好的测序文库用Illumina HiSeqTM2000 进行测序。
1.2 数据的拼接和组装
我们使用短初始获得的序列(reads)组装软件Trinity[15]做转录组从头组装Trinity 首先将具有一定长度overlap 的初始序列连成更长的片段,这些通过reads overlap 关系得到的不含空位的组装片段我们称之称为Contig。然后,我们将初始序列比对回这些组装片段,通过paired -end reads 能确定来自同一转录本的不同初步组装序列以及这些序列之间的距离,Trinity 将这些序列连在一起,得到Scaffold。最后得到的不含空位,两端不能再延长的序列,我们称之为Unigene。然后对不同样品组装得到的不含空位的序列通过序列聚类软件做进一步序列拼接和去冗余处理,得到尽可能长的非冗余不含空位的序列。最后,将不含空位的序列与蛋白数据库nr、Swiss -Prot、KEGG 和COG 做blastx 比对(<0.000 01),取比对结果最好的蛋白确定不含空位的序列的序列方向。如果不同库之间的比对结果有矛盾,则按nr、Swiss-Prot、KEGG 和COG 的优先级确定不含空位的序列的序列方向,跟以上4 个库皆比不上的编码基因则利用软件ESTScan[16]确定序列的方向。
2 结果与分析
2.1 膜果麻黄测序统计
膜果麻黄转录组测序统计结果共得到12 999 122初始序列,序列总长达到1 169 920 980 bp,初步组装得到序列数量为78 409 条,编码基因数量为49 449条。组装序列的Q20 值为95.76%,序列碱基GC 量为45.71%。
2.2 膜果麻黄测序数据初步分析
初始序列组装获得的序列长度通常是显示组装质量的指标之一,如表1可知,膜果麻黄初步组装序列的平均长度为351 bp,N50 长度(设所有初步组装序列的长度总和为x,将所有初步组装序列按序列按长度从小到大排序,并从第一条序列开始累加计算总长,当长度达到x/2 时对应的那条初步组装序列的长度即为N50)为548 bp,通过组装得到的初步组装序列长度主要分布在200~500 bp,占所有总量的78.46%;初步组装序列长度在500~1 000 bp 的序列数量为12 332,占所有总量的15.73%;长度为1 000~1 500 bp 的初步组装序列数量占总量的4.45%;长度为1 500~2 000 bp 的初步组装序列数量为835 个,占总数量的1.06%,长度大于2 000 bp 的初步组装序列数量占总数的0.29%。而不含空位的序列N50 值为663 bp,平均长度为517 bp。不含空位的序列长度分布在200~500 bp 的序列所占总量比例为62.61%;长度分布在500~1 000 bp 序列有13 313 条,所占比例为26.92%;长度在1 000~1 500 bp 的序列占总量之和的7.89%;不含空位的序列长度在1 500~2 000 bp 序列有997 条;而不含空位的序列长度分布大于2 000 bp 序列所占比例为最少的0.59%。
对组装好的膜果麻黄转录组进行的不含空位的序列的位置分布分析可得图1,由图可知不含空位的序列3’端的初始序列数量比较少,而同时不含空位的序列与其它部位匹配得到了较多的初始序列,并且其分布都较为均匀。
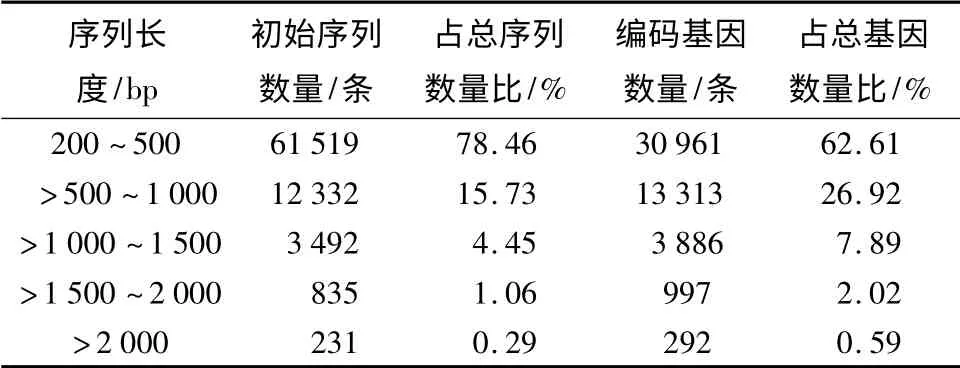
表1 膜果麻黄组装质量统计
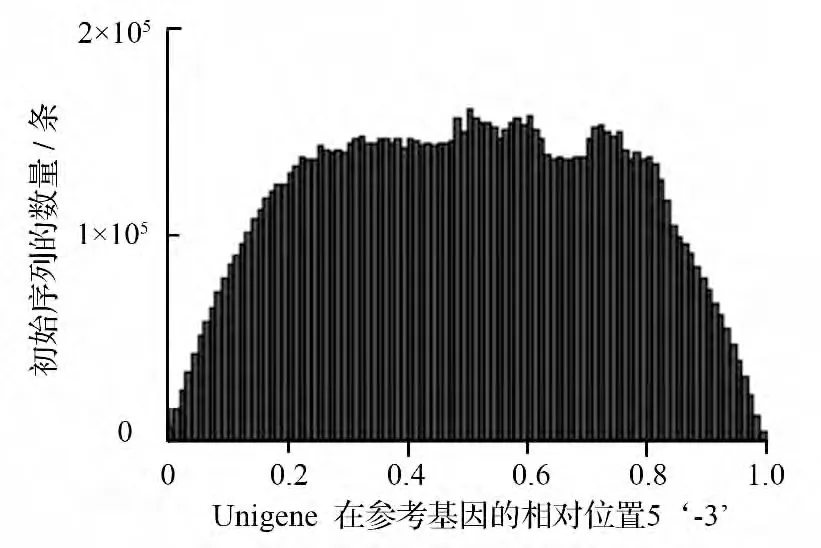
图1 初始序列在中麻黄不含空位的序列的测序随机性结果
2.3 膜果麻黄的GO 分类分析
Gene Ontology(GO)是一个国际标准化的基因功能分类体系,提供了一套动态更新的标准词汇表来全面描述生物体中基因和基因产物的属性。我们根据nr 注释信息,使用Blast2GO 软件[17]得到不含空位的序列的GO 注释信息。得到每个不含空位的序列的GO 注释后,我们用软件WEGO[18]对所有不含空位的序列做GO 功能分类统计,从宏观上认识该物种的基因功能分布特征。我们对获得的不含空位的序列注释到3 大板块:生物过程(biological process)、细胞组分(cellular component)和分子功能(molecular function)。其中生物过程占45.74%,细胞组分和分子功能分别占57.77% 和25.84%。其中下级分类中,细胞(9674,19.51%)、细胞组分(8 829,17.85%)、细胞器(7 189,14.54)、催化活性(6 376,12.89%)、代谢过程(5 252,10.62%)、结合作用(5 232,10.58%)、细胞过程(5 191,10.5%)所占的比例较大。
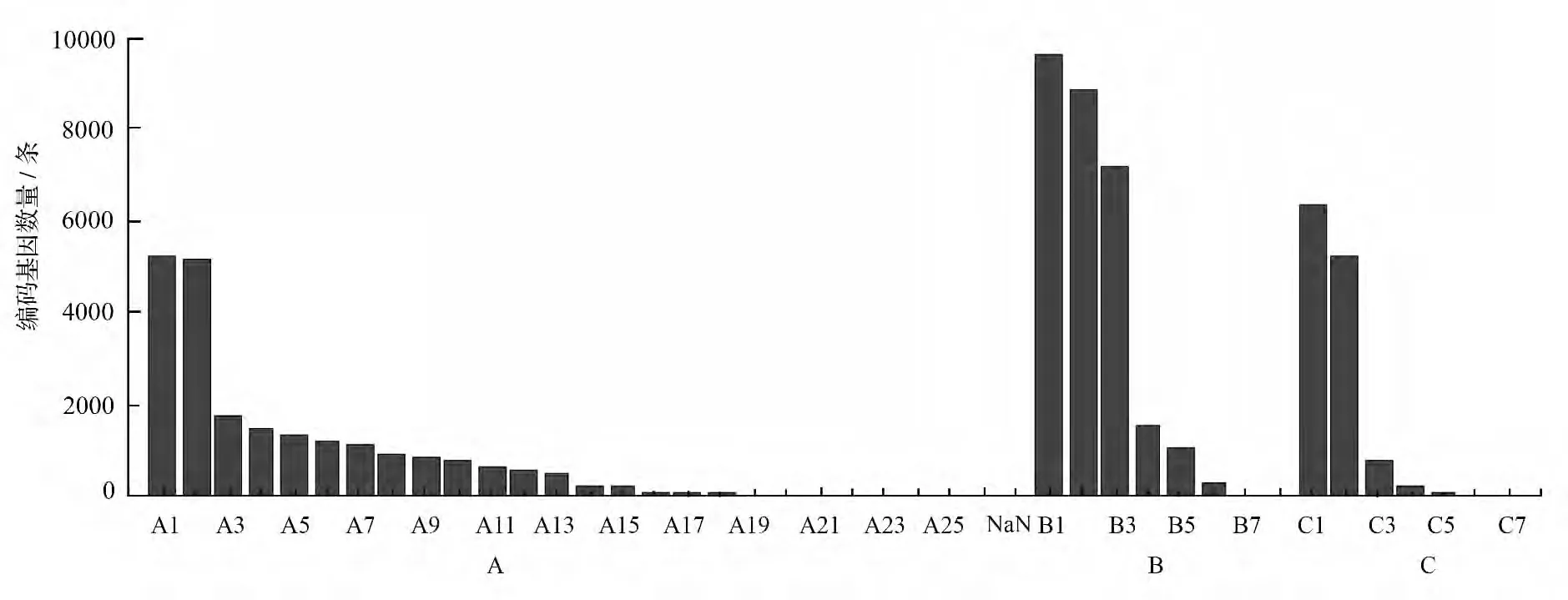
图2 膜果麻黄转录组不含空位的序列GO 分类结果
2.4 膜果麻黄的COG 分类分析
将膜果麻黄转录组所得的编码基因与COG(Cluster of Orthologous Groups of proteins)数据库进行比对,然后对其做功能分类和统计,共获得了17 751 COG 功能注释,分为26 个功能分类(图3)。从功能分类结果来看,R 类常规功能预测(General function predietion only)的编码基因数量为最多(2 861 个),占总注释数的16.12%;然后,依次是K 类转录(Transcription)(1 489,8.39%)、O类翻译后修饰(Posttranslational modification,protein turnover,chaperones)(1 452,8.18%)、L 类(Replication,recombination and repair)(1 413,7.96%)、J类(Translation,ribosomal structure and biogenesis)(1 313,7.4%)和G 类(Carbohydrate transport and metabolism)(1 093,6.16%)所占比例比较大;而Y 类(Nuclear structure)和W 类(Extracellular structures)注释最少,分别仅有5 和3 条编码基因。
2.5 膜果麻黄的KEGG 注释
KEGG 作为全球影响力最大的生物代谢数据库之一[19],涉及系统信息、基因组信息和化学信息等[20]。通过KEGG 注释发现,膜果麻黄种子萌发期间共有16 748 个编码基因参与了125 个代谢通路,排名前15 位的代谢通路见表2。其中代谢途径(Metabolic pathways)参与基因最多,达3 768 个,占所有编码基因的22.5%;然后是次生代谢产物生物合成途径(Biosynthesis of secondary metabolites)有1 888 个编码基因参与,占11.27%;再次是植物激素信号传导(Plant hormone signal transduction)的编码基因占4.54%(761 个)等。
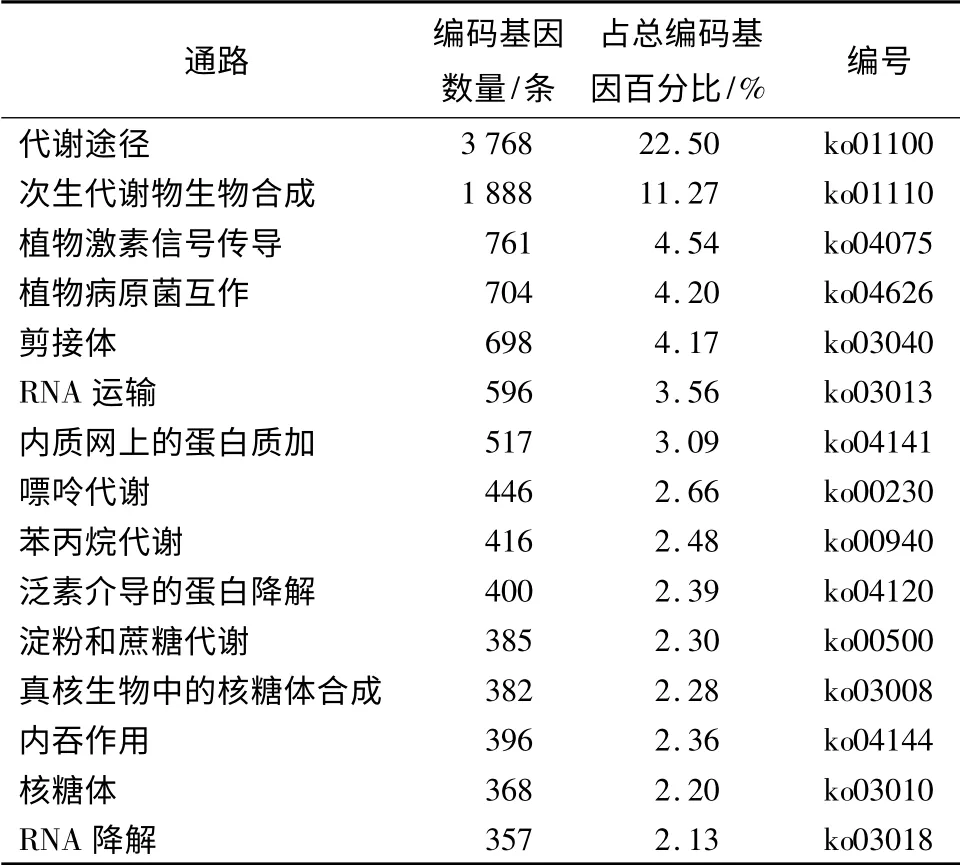
表2 KEEG 注释中数量前15 个通路
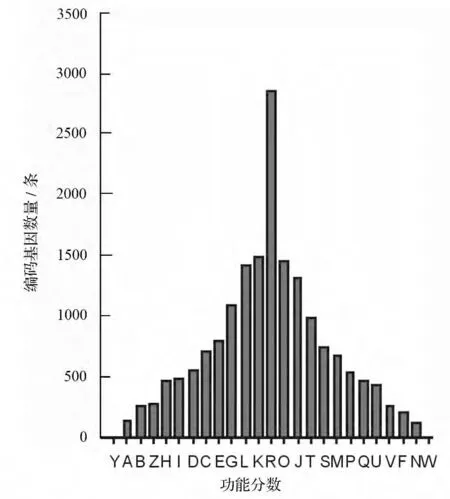
图3 编码基因的COG 分类结果
2.6 膜果麻黄芪类化合物等合成代谢途径发掘
膜果麻黄传统上认为没有药用价值,仅作为荒漠恢复灌木。KEGG 代谢途径分析显示膜果麻黄萌发种子的转录组中存在芪类、二芳基庚烷和姜醇(Stilbenoid,diarylheptanoid and gingerol biosynthesis)的合成途径。转录组中涉及该途径的基因共有230条编码基因,占所有片段总数的1.37%。由表3可看出,膜果麻黄中涉及上述合成途径的基因及其编码的酶有奎宁酸香豆酰转移酶(shikimate O -hydroxycinnamoyltransferase,HCT)、肉桂酸4 一羟基化酶(trans-cinnamate 4 -monooxygenase,C4H)、香豆酸一3 一羟基化酶(coumaroylquinate(coumaroylshikimate)3’-monooxygenase,C3’H)和咖啡酰辅酶A-O-甲基转移酶(caffeoyl-CoA O-methyltransferase,CCOAMT)。
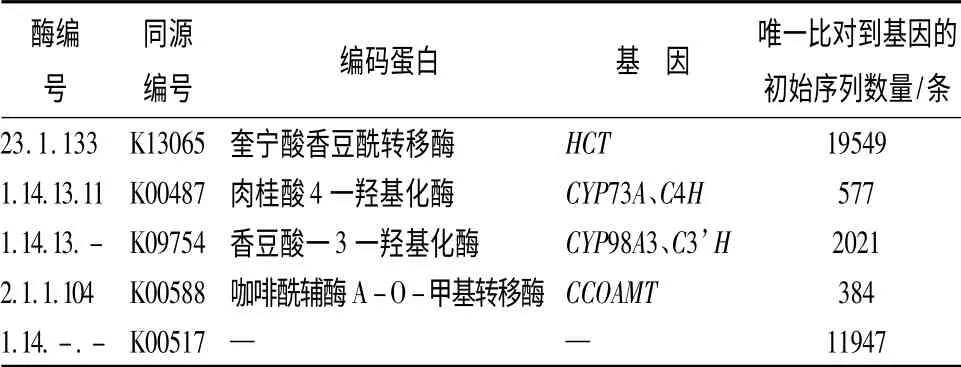
表3 膜果麻黄中涉及芪类、二芳基庚烷和姜醇合成途径的基因信息
3 结论与讨论
荒漠植物种子萌发是一个复杂的过程,荒漠植物都在生长期的进化历程都演化出了与严酷生境相适应的萌发对策[21],膜果麻黄作为西北地区重要的生态灌木之一,通过研究其种子萌发过程,对深入研究其系统进化、环境的适应及次生代谢等具有重要意义。本研究为了更加系统和完整地获取膜果麻黄种子萌发过程中的转录组数据信息,选用了不同萌发时间的膜果麻黄种子以获取覆盖度广且较为准确的信息。测序和组装结果获得了12 999 122 个初始序列,总长为1 169 920 980 bp,初步组装序列的平均长度为351 bp,N50 长度为548 bp;编码基因的N50值为663 bp,平均长度为517 bp。与前人研究相比较,如根腐线虫转录组[22]中共有326 971 条初始序列,初步组装序列的平均长度为458 bp;日本三角涡虫转录组[23]中拼接得到37 218 条编码基因数据,平均长度468 bp。这说明本研究测序所得到的数据量大,组装效果较好。
芪类次生代谢物具有抗病、抗氧化、抗肿瘤、抗炎症等多种生物活性[24],已在豆科(Leguminosae)、松科(Pinaceae)、葡萄科(Vitaceae)和买麻藤科(Gnetaceae)等不同植物中发现;二芳基庚烷具有抗肿瘤活性,而姜醇在调味品、保健食品、药品等行业应用广泛。本研究发现了有关芪类、二芳基庚烷和姜醇合成的相关230 条编码基因,其中所发现的基因大多都已在别的物种中有过克隆等其他研究。如HCT 基因是木质素生物合成途径中的一种酶,在苯丙烷C3 羟基化作用的上、下游起着双重调节作用[25-27];王雪霞[28]通过研究认为该基因对于植物的分子进化及植物分类上可能具有一定的意义;而研究表明C3H 催化苯丙烷苯环C。发生羟基化反应可能发生在香豆酸或香豆酸莽草酸/奎宁酸的水平上,虽然其最终地位还未最终确定,但该遗传特性的研究可能对C3H 在植物的进化意义及植物体的综合利用提供理论依据[29];而C4H 基因属于CYP73 亚家族,参与植物中的许多天然代谢产物如苯丙烷类、生物碱等的生物合成[30]。由于C4H 基因在植物的次生代谢中的重要作用,因此受到关广泛关注;目前已报道从杨树(Populus sp.)、亚洲棉(Goss.pium arboretum Linn.)等植物中克隆了该基因,通过对该基因的研究可以对根分泌的化感物质进行调控。
由于麻黄为非模式植物,可供参考的遗传信息相对较少,因此对其特异性的新基因的发掘还有待进一步的深入研究。以本研究所获得的转录组为基础,今后可进一步开发麻黄属的分子标记,从而对西北地区麻黄植物的遗传的结构和多样性开展研究,评估和保护其遗传资源;还能够克隆麻黄中的重要生物活性成分的合成关键基因,更好利用其药用价值;同时也为麻黄这种荒漠植物种子萌发过程中抗逆功能基因的发掘及其优良性状遗传改良等提供了大量的遗传数据资源。
[1] 刘媖心.中国沙漠植物志:3 卷[M].北京:科学出版社,1992.
[2] 孔红,陈荃,马骥.麻黄属2 种植物的核型研究[J].兰州大学学报:自然科学版,2001,37(3):100 -103.
[3] 张盹明,杨自辉,王继和,等.2 种麻黄光合及其耐逆性分析[J].西北植物学报,2007,27(7):1473 -1478.
[4] Huang J,Giannasi D E,Price R A.Phylogenetic relationships in Ephedra (Ephedraceae)inferred from chloroplast and nuclear DNA sequences[J].Molecular Phylogenetics and Evolution,2005,35(1):48 -59.
[5] Groff G W,Clark G W.The botany of Ephedra in relation to the yield of physiologically active substances[M].San Francisco:University of California Press,1928.
[6] Caveney S,Charlet D A,Freitag H,et al.New observations on the secondary chemistry of world Ephedra (Ephedraceae)[J].American Journal of Botany,2001,88(7):1199 -1208.
[7] Gutterman Yitzchak.Survival strategies of annual desert plants[M].Berlin:Springer-Verlag Berlin and Heidelberg GmbH,2002.
[8] Schuster S C.Next-generation sequencing transforms today’s biology[J].Nature,2007,200(8):16 -18.
[9] Ansorge W J.Next-generation DNA sequencing techniques[J].New Biotechnology,2009,25(4):195 -203.
[10] Huang W,Marth G.EagleView:a genome assembly viewer for next-generation sequencing technologies[J].Genome Research,2008,18(9):1538 -1543.
[11] Gahlan P,Singh H R,Shankar R,et al.De novo sequencing and characterization of Picrorhiza kurrooa transcriptome at two temperatures showed major transcriptome adjustments[J].BMC Genomics,2012,13:126.
[12] Sultan M,Schulz M H,Richard H,et al.A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome[J].Science,2008,321:956 -960.
[13] Wang E T,Sandberg R,Luo S,et al.Alternative isoform regulation in human tissue transcriptomes[J].Nature,2008,456:470-476.
[14] 吴琼,孙超,陈士林,等.转录组学在药用植物研究中的应用[J].世界科学技术:中医药现代化,2010(3):457 -462.
[15] Grabherr M G,Haas B J,Yassour M,et al.Full-length transcriptome assembly from RNA-Seq data without a reference genome[J].Nature Biotechnology,2011,29(7):644 -652.
[16] 张楠,孙桂玲,戴均贵,等.银杏细胞转录组高通量测序及分析[J].中国生物工程杂志,2013,33(5):112 -119.
[17] Conesa A,Götz S,Garcia G J M,et al.Blast2GO:a universal tool for annotation,visualization and analysis in functional genomics research[J].Bioinformatics,2005,21(18):3674 -3676.
[18] Ye J,Fang L,Zheng H,et al.WEGO:a web tool for plotting GO annotations[J].Nucleic Acids Research,2006,34(增2):293 -297.
[19] Kanehisa M,Goto S,Sato Y,et al.KEGG for integration and interpretation of large-scale molecular data sets[J].Nucleic Acids Res,2012,40:109 -114.
[20] 周国艳,胡望雄,徐建红,等.整合多个组学(omics)分析植物代谢产物及其功能[J].浙江大学学报:农业与生命科学版,2013,39(3):237 -245.
[21] 张勇,薛林贵,高天鹏,等.荒漠植物种子萌发研究进展[J].中国沙漠,2005,25(1):106 -112.
[22] Haegeman A,Joseph S,Gheysen G.Analysis of the transcriptome of the root lesion nematode Pratylenchus coffeae generated by 454 sequencing technology[J].Molecular and Biochemical Parasitology,2011,178(1):7 -14.
[23] Qin Y,Fang H,Tian Q,et al.Transcriptome profiling and digital gene expression by deep-sequencing in normal/regenerative tissues of planarian Dugesia japonica[J].Genomics,2011,97(6):364 -371.
[24] 何水林,郑金贵,林明,等.植物芪类次生代谢物的功能、合成调控及基因工程研究进展[J].农业生物技术学报,2004,12(1):102 -108.
[25] Hoffmann L,Maury S,Martz F,et al.Purification,cloning,and properties of an acyltransferase controlling shikimate and quinate ester intermediates in phenylpropanoid metabolism[J].J Biol Chem,2003,278(1):95 -103.
[26] Raes J,Rohde A,Christensen J O R H,et al.Genome-wide characterization of the lignification toolbox in Arabidopsis[J].Plant Physiology,2003,133(3):1051 -1071.
[27] Reddy M S,Chen F,Shadle G,et al.Targeted down-regulation of cytochrome P450 enzymes for forage quality improvement in alfalfa (Medicago sativa L.)[J].Proceedings of the National Academy of Sciences of the United States of America,2005,102(46):16573 -16578.
[28] 王雪霞,曹方,薛永常.杨树木质素合成酶hct 基因的克隆及核苷酸序列分析[J].辽宁林业科技,2009(6):4 -7.
[29] 聂会忠,薛永常.杨树木质素合成酶c3h 基因的克隆及其序列分析[J].西北植物学报,2008,28(5):889 -894.
[30] Schuler M A.Plant cytochrome P450 monooxygenases[J].Crit Rev Plant Sci,1996,15:235 -284.
