基于改进标记传播算法的基因表达谱数据研究
2015-03-03葛芳郭有强王磊马程
葛芳,郭有强,王磊,马程
(蚌埠学院 计算机科学与技术系 安徽 蚌埠 233030)
基于改进标记传播算法的基因表达谱数据研究
葛芳,郭有强,王磊,马程
(蚌埠学院 计算机科学与技术系 安徽 蚌埠 233030)
提出一种改进的标签传播算法,并将其应用于基因表达谱数据分析中.首先使用概率矩阵表示基因表达数据,将少量样本标记为已知,同时定义一个标记序列表示样本的类别属性;然后通过迭代公式更新标记序列,得到标记序列的收敛解,并证明了该收敛解的唯一性;最后采用正负标记的方式,根据标记序列各分量的符号差异实现数据类别的划分.经过癌症数据集实验的验证,证明了提出的方法可以快速有效地实现基因表达数据的聚类.
半监督学习;概率转移矩阵; 标记传播; 基因表达谱数据
DNA微阵列技术为肿瘤学提供了一种全新的研究手段.一次基因芯片实验可以获得成千上万个基因的信息,而随着DNA微阵列技术的进步和仪器设备的更新,基因表达谱数据将不断积累,这类数据的主要特点是样本少、维数高、冗余基因和噪声多,因此,如何对这类“新型”数据进行分析,挖掘其中包含的有效信息,成为生物信息学研究的重点课题.
传统的基因表达谱分析方法主要是对基因数据进行信息基因选取或特征属性提取后,再用相应的分类或聚类方法对样本进行识别[1-2].其中,聚类是一种无监督学习方法,传统的聚类方法,如K均值[3]、模糊C均值[4]和自组织映射[5]等,都要求数据呈球状分布,且这些算法易陷入局部最优.禤世丽[6]等人针对上述问题,提出了基于粒子对(PPO)与差分进化(DE)混合算法,并结合K-means进行聚类.近年来,基于图划分理论的聚类方法成为机器学习领域的研究热点.2001年,Meila和Shi[7]把图的节点的相似性解释为马尔科夫链中的随机游走,同时分析了这种随机游走的概率转移矩阵的特征向量,并提出了多个特征相似矩阵组合下的谱聚类算法;2006年,Zhu[8]用高斯随机场的方法处理半监督学习问题,提出了标记传播算法,并根据随机游走对标记传播算法进行了概率解释;2010年,Bai等人[9]将标记传播进行了扩展,提出了基于上下文分析的图传导算法,并将其应用于图像分割、形状检索和匹配等领域.
标记传播是一种基于图的半监督学习方法,它利用少量已标记类别的样本,通过传播标记的方式来识别未知类别的样本.标记传播算法能在任意形状的样本空间中进行聚类,克服了传统聚类方法易陷入局部最优的缺点,但原始标记传播算法迭代次数过大,每次迭代前都要重新标记已知类别的样本,且最终的划分准则也并不明确.针对上述问题,提出了一种改进的LP算法,并应用于实际的癌症数据的分析中.癌症数据集的实验结果证明了该方法的有效性.
1 基因表达数据的概率转移矩阵表示

(1)
其中,aij表示基因gj在样本xi中的表达值.
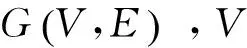
(2)
其中,dij表示节点xi和xj之间的欧氏距离,σ是权重参数,通常,σ可以根据节点xi和xj的K个近邻的平均距离来自适应调整大小[10],K是一个经验值.显然,对于任意的xi,xj和σ,有0≤Wij≤1,反映了节点xi和xj之间的亲近程度,Wij越大,则节点xi和xj越有可能属于同一类.假设xi是一个已标记的节点,那么从节点xi转移到节点xj的概率为
(3)
其中,di=∑kWik为节点xi的度,表示与其连接的所有节点的边权之和.因此,概率转移矩阵可表示为
P=D-1W
(4)
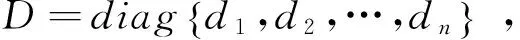
2 标记传播算法
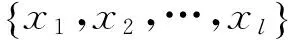
标记传播的步骤如下:
Step1: 传播标记,f=Pf;
Step2: 强化标记数据,fL=YL;
Step3: 返回Step1,直到f收敛.
通过第一步,将已标记样本节点的标记传播至相邻的节点;此外,在标记的传播过程中,为保证传播过程的正确性,已知样本的标记值不变,用以保持标记数据的强度;算法迭代终止后,标记序列f包含了样本的类别信息.文献[8]给出了标记传播算法的收敛解
(5)
其中,PUU和PUL是概率转移矩阵P的子矩阵块,即
(6)
因此,标记传播算法的解将收敛于一个固定解,需要指出的是,根据式(5)可以直接求出未知样本的标记.
3 改进的标记传播算法
通过上述方法任意数据点xi都可以映射为相应的实数值fi,但本文算法在每次迭代前都要重新标记已知样本,且由于标记传播过程和标记强化过程的分离,造成算法迭代次数过大;同时,本文算法采用0-1标记,需要选取适合的阈值对样本进行分类,Zhu等人[8]使用的0.5阈值缺乏稳定性,应用于复杂数据时并不能取得很好的效果.
3.1 改进的标记传播算法
令α表示标记传播和标记强化的平衡参数,且满足0<α<1,反映了数据点从其近邻点获取的标记信息的比例,将参数α加入迭代公式,在t+1次迭代后,数据点xi的标记为
(7)
其中,f0=(YL,0,…,0),在每次迭代过程中,数据点从已标记节点中获取一部分标记信息;同时,由于相似节点间的权重较大,数据点可以从其近邻中获取一部分标记信息,当传播终止后,相似节点的分布情况也趋于一致.因此,迭代公式可以改写为
(8)
利用式(8)来更新每个数据的标记,直至收敛.这里,并不采用原始LP算法中的0和1标记,而采用正负标记的方式,如对于两类问题,将其中一类的若干个样本标记为1,而另一类的若干个样本标记为-1,对于最终的收敛结果,就能以零为分割点对未知类别的样本进行划分.
3.2 收敛性证明
本节将证明利用上述算法得到的f是收敛的,由式(8)可以得到是收敛的,由式(8)可以得到
(9)
由于Pij>0且∑jPij=1,根据Perron-Frobenius定理[11],矩阵P的谱半径小于1;同时,由于0<α<1,因此
(10)
(11)
其中,I为n×n的单位矩阵,因此,ft将收敛于
(12)
因此,本文算法的解也是收敛的.同样,可以根据式(12)直接求出未知样本的标记.
4 实验结果与分析
4.1 实验数据
选用两组公开的癌症数据集:白血病数据(leukemia),共52个样本,其中24个样本被确定为急性淋巴白血病(ALL),28个被确定为急性粒细胞白血病(AML),每个样本含有12564个基因表达数据;结肠癌数据(coloncancer),共62个样本,其中22个样本被确定为正常样本,40个被确定为肿瘤样本,每个样本含有2000个基因表达数据.实验是在酷睿双核主频2.60GHz,内存2G的计算机上运行的.
癌症基因表达谱数据的获取过程十分复杂,所得到的数据含有大量的噪声,同时每个样本都记录了组织细胞中所有可测基因的表达水平,但只有较少数基因包含与类别相关的信息,因此,对数据进行预处理是必要的,定义下式对基因表达谱数据进行筛选
(13)
其中,max(i),min(i)和mean(i)分别表示第i个基因于所有样本当中表达水平的最大值,最小值和平均值.T为给定的阈值.如果某个基因的表达情况符合式(13)便将该基因剔除.
4.2 白血病数据实验
将白血病数据的第1号样本(ALL)标记为1,第52号样本(AML)标记为-1,分别进行5、10、50、200次迭代,更新每个样本点的标记,观察标记序列各分量的变化,结果如图1所示:
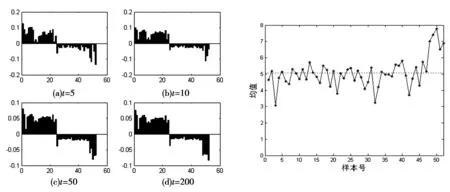
图1 白血病数据的标记传播过程 图2 白血病数据样本均值
图1中,标记序列的前24个分量对应的是白血病数据集的ALL样本,后28个分量对应AML样本.从图1中可以看到,当进行5次迭代后,ALL样本的标记都大于零,而AML样本的标记都小于零,所有样本被正确的划分为两类,说明该方法可以快速且有效的实现样本类别的划分;同时,图1(c)和(d)变化不明显,即表示较少次数的迭代便可得到最终结果.另外,ALL样本中,第3号样本的分布明显不同于其他样本,AML样本中,第48、49、50、51和52号样本也不同于其他样本,这是由数据的分布特点决定的,图2是白血病数据集中各样本均值.
4.3 结肠癌数据实验
将结肠癌数据的第1号样本(正常组织样本)标记为1,第62号样本(肿瘤组织样本)标记为-1,同样分别进行5、10、50、200次迭代,结果如图3所示.
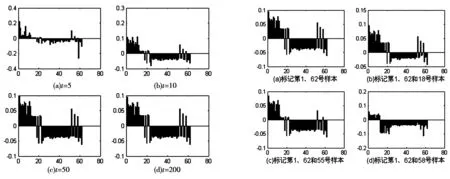
图3 结肠癌数据的标记传播过程 图4 异常样本被标记为已知的迭代结果
图3中,标记序列的前22个分量对应的是结肠癌数据集的正常组织样本,后40个分量对应肿瘤组织样本.从图3中可以看到,当经过5次迭代后,两类样本标记的区别并不明显,但经过10次迭代后,就可以看出两类样本的标记的差异性,在正常组织样本中,第18和20号样本被错误标记,而在肿瘤组织样本中,第52、55和58号样本被错误标记,分类准确率达到91.94%,实际情况中,这些样本中含有较多的偏离值和异常点;同时,进行50次和200迭代后,样本标记的变化已经不明显,同样说明了该方法可以快速实现标记序列的收敛.
为了观察初始标记点对最终迭代结果的影响,分别将结肠癌数据中被错误标记的第18、55和58号样本标记为已知,结果如图4所示.
图4(a)中仅标记了第1和62号样本;图4(b)除标记第1和62号样本外,同时将第18号样本标记为1;图4(c)、(d)除标记第1和62号样本外,分别将第55号和58号样本标记为-1.由图4(b-c)可知,当将第18或55号样本标记为已知,那么与第18或55样本相似的样本,其标记值也趋向于与第18或55样本相似(实际表现为样本标记值的绝对值减小).由图4(d)可知,当将第58号样本标记为已知,第13至22号样本全部被错误标记,同时第1至12号样本的标记值的绝对值减小,这同样是由于在标记传播过程中,某个样本的标记是根据其近邻样本点的标记进行更新的.因此得出结论,在试验中,若把异样表达的样本标记为已知,与该样本表达相似的样本趋向于已标记的异样表达样本,便会出现样本被错误标记的现象.
4.4 对比验证
为进一步验证该方法的正确性,设计了两组对比实验:
第一组实验:将该方法与局部线性嵌入(LLE)算法[12-13]和NJW谱聚类算法[14]进行对比.LLE是一种非线性特征提取算法,先以LLE进行特征提取,再用KNN对样本进行分类;NJW算法采用规范化相似矩阵的前K个最大特征值对应的特征向量作为数据的特征表示,再用K均值等对特征空间的样本点进行聚类.结果如表1所示:

表1 本文方法与基于图的方法的实验结果比较
由表1可知,从准确率方面,本文方法明显优于另外两种基于图的方法,LLE算法是一种有效的可视化方法,但要求数据服从流形分布,其提取的特征并不具有很强的分类能力;NJW算法属于无监督学习,提取的特征向量未必能反映数据结构;而本文方法利用已知样本的类别标记对未知样本的标记进行传播,在传播过程中,某个样本的标记是根据其近邻样本点的标记进行更新的,最后得到的收敛结果也更符合原始数据分布的特点.同时,在时间效率上,本文方法并不具有明显的优势,这是因为这几种方法都是利用构图的方法将高维样本映射到低维空间,构造的矩阵规模是相同的.
第二组实验:将本文方法与传统的S2N_KNN法[1]和CLUSTER_S2N[4]进行对比,S2N_KNN以“信噪比”为指标选取特征基因,再用K-近邻(KNN)分类器对样本进行分类;CLUSTER_S2N先用K均值将数据聚类,再以“信噪比”选取特征基因,最后用支持向量机(SVM)实现样本的分类.结果如表2所示:

表2 本文方法与传统方法的实验结果比较
由表2可知,无论从准确率还是运算效率等方面,本文方法都优于另外两种传统方法,由于基因之间普遍存在相关性,传统方法以降维来提取特征基因势必会丢失部分含有分类信息的基因,而本文方法将样本数据作为高维空间中的点,所构造的概率转移矩阵反映了样本之间的关系,使得原来的样本分类信息完全映射到低维的概率转移矩阵中.同时,在时间效率上,提出的方法同样优于后两种方法,这是因为传统方法在前期处理中对高维数据进行复杂运算,进行一次甚至多次降维处理,而提出的方法首先以样本为节点构图,其低样本特性决定了构造的矩阵规模较小,从而具有较低的运算复杂度,有利于基因表达数据的快速分类.
5 结 语
基于图的半监督学习是当前模式识别领域的研究热点.提出一种改进的标记传播算法,本文算法通过引入标记传播过程和标记强化过程的平衡参数,克服了传统标记传播算法迭代次数过大和重复标记数据点的缺点;同时在数据类别划分时使用正负标记的方式,避免了采用0-1标记时阈值选取的不确定性.通过癌症数据的实验,证明了该方法可快速且有效地实现基因表达谱数据的聚类.
[1]GULOBTR,SLONIMDK,TAMAYOP,etal.Molecularclassificationofcancer:classdiscoveryandclasspredictionbygeneexpressionmonitoring[J].Science, 1999, 286(5439):531-537.
[2]KANCHERLAK,MUKKAMALAS.FeatureselectionforlungcancerdetectionusingSVMbasedrecursivefeatureeliminationmethod[J].MachineLearningandDataMininginBioinformatics, 2012, 7246:168-176.
[3] 禤浚波, 吴小霞, 王珍珍,等. 基于粒子对和极值优化的基因聚类混合算法研究[J]. 计算机应用研究, 2011, 28(10):3675-3677,3680.
[4]TARIL,BARALC,KIMS.Fuzzyc-meansclusteringwithpriorbiologicalknowledge[J].JournalofBiomedicalInformatics, 2009, 42(1):74-81.
[5]PATTERSONAD,LIH,EICHLERGS,etal.UPLC-ESI-TOFMS-basedmetabolomicsandgeneexpressiondynamicsinspectorself-organizingmetabolomicmapsastoolsforunderstandingthecellularresponsetoionizingradiation[J].AmericanChemicalSociety, 2008, 80(3):665-674.
[6] 禤世丽,杨秋叶,张超英,等. 基于粒子对和差分进化的基因表达数据聚类[J]. 计算机应用研究, 2012, 29(7):2484-2487.
[7]MELIAM,SHIJin-bo.Learningsegmentationbyrandomwalks[J].InAdvancesinNeuralInformationProcessing,2000, 10(2):873-879.
[8]ZHUXiao-jin.Semi-Supervisedlearningwithgraphs[D].Doctoraldissertation,CarnegieMellonUniv,CMU-LTI-05-192,2005.
[9]BAIXiang,YANGXing-wei,LATECKILJ,etal.Learningcontext-sensitiveshapesimilaritybygraphtransduction[J].IEEETransactionsonPatternAnalysisandMachineIntelligence, 2010, 32(5):861-874.
[10]ZELNIK-MANORL,PERONAP.Self-tuningspectralclustering[J].AdvancesinNeuralInformationProcessingSystems, 2004, 17(2):1601-1608.
[11]GOLUBGH,VANLOANCF.Matrixcomputatio[M].Baltimore:TheJohnsHopkinsUniversityPress, 1996.
[12]PILLATIM,VIROLIC.Supervisedlocallylinearembeddingforclassication[A].anapplicationtogeneexpressiondataanalysis[C].Proceedingsof29thAnnualConferenceoftheGermanClassicationSociety(GfKl2005), 2005.15-18.
[13]ZHAOLing-xiao,ZHANGZheng-yue.Supervisedlocallylinearembeddingwithprobability-baseddistanceforclassification[J].ComputersandMathematicswithApplications, 2009, 57: 919-926.
[14]NGAY,JORDANMI,WEISSY.Onspectralclustering:analysisandanalgorithm[C].DIETTERICHTG,BECKERS,GHAHRAMANIZ.AdvancesinNeuralInformationProcessingSystems,Cambridge,MA:MITpress, 2002,849-85.
[责任编辑:王军]
The analysis of gene expression profiles based on improved label propagation algorithm
GE Fang,GUO Youqiang, WANG Lei, MA Cheng
(Department of Computer Science and technology, Bengbu College, Bengbu 233030, China)
In this paper, an improved label propagation algorithm was proposed and introduced into the analysis of gene expression profiles. First, the probability transition matrix was constructed with gene expression profiles. Meanwhile, the label sequence which indicates the class information was defined and several samples were marked as labeled data. Then, the label sequence was updated by an iterative formula and the convergence solution of the label sequence was obtained, which was proved to be the unique solution. Finally, the clustering problem was solved by using plus-minus label which was on the basis of the signs of the label sequence. Experiments on the cancer data demonstrate our method is feasible and effective.
semi-supervised learning; probability transition matrix; label propagation; gene expression profile data
2015-03-02
安徽省自然科学基金资助项目(No.11040606M151);蚌埠学院自然科学基金资助项目(No.2014ZR26,No.2013ZR06)
葛芳(1986-), 女, 安徽亳州人, 蚌埠学院助教, 硕士研究生, 主要从事模式识别、计算生物学的研究; 郭有强(1966-), 男, 蚌埠学院教授,博士, 硕士生导师, 主要从事为数据挖掘、机器学习的研究.
TP 18
A
1672-3600(2015)06-0063-06
