血管瘤样纤维组织细胞瘤的临床病理观察
2015-02-23焦南林卢林明武秀秀徐国祥
焦南林,卢林明,张 帆,武秀秀,徐国祥
(1. 皖南医学院第一附属医院 弋矶山医院 病理科,安徽 芜湖 241001;2. 皖南医学院 病理学教研室,安徽 芜湖 241002)
·临床医学·
血管瘤样纤维组织细胞瘤的临床病理观察
焦南林1,卢林明2,张 帆1,武秀秀1,徐国祥1
(1. 皖南医学院第一附属医院 弋矶山医院 病理科,安徽 芜湖 241001;2. 皖南医学院 病理学教研室,安徽 芜湖 241002)
目的:探讨血管瘤样纤维组织细胞瘤的临床病理特征、分子遗传学改变、病理诊断及鉴别诊断。方法:收集2例血管瘤样纤维组织细胞瘤,复习相关文献,分析其临床病理学特征。采用免疫组织化学和荧光原位杂交方法分别观察其免疫表型和EWSR1基因重排情况。结果:2位患者年龄为9岁和24岁,分别表现为左上臂和颈前区包块。大体上,肿瘤界限清楚、直径1.5~2.0 cm。镜下,瘤组织周边见淋巴细胞、浆细胞浸润带,并被一层较厚的纤维性假包膜包绕;瘤细胞梭形和胖梭形,异型性轻到重度,可见多核巨细胞,核分裂像可见。其中1例瘤细胞巢内含有出血性囊腔,另一例为实体型。免疫组织化学显示瘤细胞均弥漫表达vimentin,部分细胞表达CD68、CD99,少量细胞表达SMA、EMA;Ki-67增殖指数为3%~10%;荧光原位杂交检测结果显示2例均存在EWSR1基因重排。2例均经手术完整切除肿块,随访54个月未见复发和转移。结论:血管瘤样纤维组织细胞瘤是少见的交界性肿瘤,多见于儿童和青少年,好发于四肢,其次位于躯干和头颈部,易与其他肿瘤混淆。熟悉这一病变的临床、病理形态特点,结合免疫组织化学和分子遗传学检测能够避免误诊。
纤维组织细胞瘤;血管瘤样;免疫表型;EWSR1
【DOI】10.3969/j.issn.1002-0217.2015.05.009
血管瘤样纤维组织细胞瘤(angiomatoid fibrous histiocytoma, AFH)又称血管瘤样恶性纤维组织细胞瘤,是一种少见的交界性软组织肿瘤,国内文献报道较少。本文收集了我院病理科2010年10月诊断的2例AFH,并复习相关文献,探讨其临床特点、病理学形态特征、免疫组织化学表型和分子遗传学改变,以期提高对该病变的诊断水平。
1 资料与方法
1.1 临床资料 例1,男性,9岁,发现左上臂皮下包块半年。例2,女性,24岁,发现颈前区皮下包块8年。包块均缓慢增大,无发热、消瘦、贫血、局部疼痛及皮肤发红等不适。
1.2 方法 标本经10%中性福尔马林固定,常规脱水,石蜡包埋,切片厚度4 μm,HE染色。免疫组织化学采用EnVision两步法,所用一抗CKpan、EMA、vimentin 、CD68、CD99、SMA、desmin、Ki-67等均购自福州迈新生物技术开发公司。荧光原位杂交(fluorescence in situ hybridization,FISH)选用安必平公司EWSR1基因断裂检测探针,操作严格按试剂盒说明书进行。
2 结果
2.1 巨检 2例病变组织均为结节样肿块,界限清楚。例1肿块直径1.5 cm,切面灰红色,实性,质地中等;例2肿块直径2.0 cm,切面灰白、灰黄,实性,质地韧。
2.2 镜检 病变均边界清晰,周边区域可见显著的慢性炎细胞浸润带,以淋巴细胞和浆细胞为主,呈套样结构,伴淋巴滤泡形成,并被一层较厚的纤维性假包膜包绕,似淋巴结,但无边缘窦(图1)。肿瘤主要由梭形和胖梭形细胞组成,呈束状或不规则排列,瘤细胞异型性由轻到重度(图2),核分裂像0~2个/10HPF,可见多核巨细胞,间质内见出血和含铁血黄素沉积。其中1例瘤细胞巢内含有多灶性的出血性囊腔,无内皮细胞内衬(图3),另1例为实体型。
2.3 免疫组织化学 瘤细胞vimentin弥漫强阳性(图4),部分细胞表达CD68和CD99,少量细胞表达SMA和EMA;其中1例少量细胞表达desmin、Calponin、bcl-2;不表达MyoD1、Myogenin、Myoglobin、CD21、CD23、CD35、CD34、ALK-1、S-100蛋白、HMB45和CKpan;CD34显示出血性囊腔无内皮细胞内衬,为假血管腔隙(图5);Ki-67增殖指数为3%~10%。
2.4 FISH 计数200个肿瘤细胞核内的荧光信号,结果显示2例病变组织中的红、绿分离信号比例均超过阴性阈值,判定存在EWSR1基因重排(图6)。
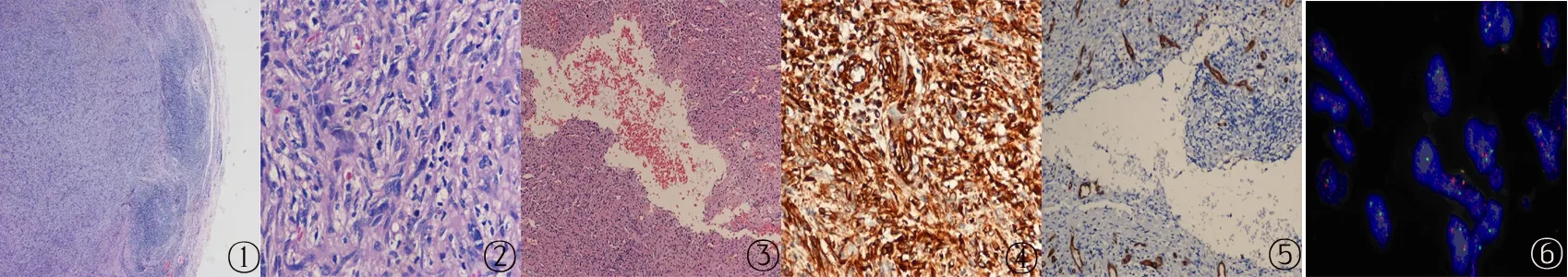
图1 肿瘤组织×40;图2 肿瘤细胞异型 ×400;图3 出血性囊腔 ×100;图4 肿瘤细胞vimentin×400;图5 出血性囊腔CD34 ×200;图6 肿瘤细胞 EWSR1基因FISH检测
2.5 治疗及随访结果 两例均经外科手术完整切除肿块,术后随访54个月未发现复发和转移。
3 讨论
AFH由Enzinger于1979年对其临床病理特征作了详细描述,最初被命名为血管瘤样恶性纤维组织细胞瘤,并作为恶性纤维组织细胞瘤(MFH)的一种亚型[1]。但随后的研究发现,其在生物学行为上呈低度恶性或中间性,少数病例可复发,偶有转移[2-4],且瘤细胞分化方向不明确,WHO分类将其划入分化未定的交界性肿瘤(WHO)[5]。本瘤较少见,国内除范钦和等报道了一组例外,其余多为个案报道,且对其分子遗传学改变少有报道[4, 6]。
3.1 临床特征 文献报道中AFH的发病年龄范围较广,从新生儿至71岁均可发病,但明显好发于儿童和青少年,中位年龄不超过20岁,女性略多见。好发部位为四肢,部分病例位于躯干和头颈部,约2/3的病变位于有正常淋巴结的区域,如肘窝、腋窝、颈前和颈后,少见的发病部位还包括肺、纵膈、女性外阴、腹膜后和卵巢[1, 3, 7]。发生在少见部位的病例患者常常年龄较大、男性略多、肿块较大、全身症状出现几率大、多为黏液型、复发率高[7]。AFH临床表现多为软组织内缓慢生长的结节状、多结节状或囊性无痛性肿块,直径0.8~12 cm。少数患者可伴有系统性症状,包括发热、贫血、消瘦和多克隆丙种球蛋白病等,肿瘤切除后症状可缓解[1-2]。影像学检查有助于术前诊断,但往往缺乏特异性,MRI显像往往显示病灶内有多发囊性区域,T1和T2WI显示病灶周边有包膜,以及可能代表含铁血黄素颗粒的磁敏感伪影[8]。
3.2 病理特征 大体上,AFH通常界限清楚,切面呈灰褐色,质地坚实,类似淋巴结,切面可见不规则的出血性腔隙。镜下组织学特征包括:①有致密的纤维性假包膜;②肿瘤周边围绕淋巴细胞浆细胞浸润带,可伴有淋巴滤泡或生发中心形成,以至于在低倍镜下容易被误诊为淋巴结;③瘤细胞巢内含有多灶性的出血性囊腔,无内皮细胞内衬,为假血管性腔隙,伴间质内出血和含铁血黄素沉积;④位于中心的肿瘤性实质细胞常为梭形或组织细胞样,呈特征性合体样排列[1, 3]。梭形细胞核染色质常细腻,可见核分裂像,但多<5个/HPF,胞质呈淡嗜伊红色。近1/5的病例中,瘤细胞显示异型性,并可见多核巨细胞[2]。另有少数病例可呈小细胞样,透明细胞样、横纹肌母细胞样,肺水肿样,黏液基质中排列成条索状等[7]。本组病例形态学改变符合上述基本特征。
AFH免疫组织化学检测无特异性诊断指标,文献显示多数病例瘤细胞表达vimentin,约半数病例瘤细胞表达desmin、EMA、CD68和CD99,不表达CD34、CD21、CD35、S-100蛋白和CKpan[3, 7, 9]。因此,免疫组织化学主要为鉴别诊断提供依据。细胞遗传学上,AFH存在3种特征性易位并形成相应的融合基因,包括:t(2:22)(q33:q12)形成EWSR1-CREB1融合基因,t(12:16)(q13:p11)形成FUS-ATF1融合基因,及t(12:22)(q13:q12)形成EWSR1-ATF1融合基因,因此,理论上,只需要EWSR1和FUS两种FISH探针就可以检测出上述三种分子遗传学改变。Kao等文献报道,FISH和逆转录多聚酶链反应(RT-PCR)显示大多数AFH病例(57/69)存在EWSR1基因重排,仅个别病例存在FUS基因重排(1/69)[ 7, 9-12]。因此,笔者首先使用EWSR1 FISH断裂探针进行检测,结果显示2例均存在EWSR1基因重排。EWSR1基因重排也存在于Ewing肉瘤/原始神经外胚层肿瘤、胃肠道透明细胞肉瘤样肿瘤、肺原发性黏液样肉瘤、经典的透明细胞肉瘤等[10, 13],而这几种肿瘤在发病部位、形态学和免疫表型上与AFH有较大的差异,因此,EWSR1基因重排有望成为AFH的诊断依据之一。
3.3 鉴别诊断 ①多形性恶性纤维组织细胞瘤/未分化高级别多形性肉瘤:虽可有囊性出血,但一般好发于老年人深部组织,组织成分复杂多样且异型性明显,可见轮辐状排列结构。缺乏AFH特征性的镜下结构特点及分子遗传学特征。②动脉瘤性纤维组织细胞瘤:是良性皮肤纤维组织细胞瘤的一个少见亚型,好发于中年成人,组织学特征是低倍镜下可见朝向肿瘤中心多个含血的腔隙,囊腔无内皮细胞内衬,常常可以见到细胞成分多样和排列成轮辐状结构等典型的纤维组织细胞瘤改变,但无AFH的特征性镜下结构特点[14]。③滤泡树突状细胞肉瘤:主要发生在淋巴结内,特别是颈部和腋下淋巴结,少数发生在淋巴结外。患者年龄14~80岁,中位年龄46岁。镜下显示特征性的双相型细胞形态,有梭形至卵圆形的瘤细胞和混杂的大量小淋巴细胞组成,部分肿瘤内血管比较明显,少数病例内可见扩张的假血管性腔隙。免疫组化瘤细胞表达CD21、CD23、CD35、CD19(IF8),小部分病例可灶区或弱阳性表达S-100蛋白、EMA、SMA和CD68[15]。④炎性肌纤维母细胞肿瘤:由分化性的纤维母细胞/肌纤维母细胞组成的中间性肿瘤,也好发于儿童和青少年,多数病例位于肺、肠系膜、大网膜和腹膜后,少数可发生于皮肤。肿瘤呈结节状或分叶状,由增生的胖梭形纤维母细胞/肌纤维母细胞组成,呈束状或旋涡状排列,间质内伴有大量的炎细胞浸润,多为成熟的浆细胞、淋巴细胞和嗜酸性粒细胞,少数为中性粒细胞,可见生发中心形成。间质可呈黏液水肿样,瘤细胞间可伴有不同程度的胶原化。免疫组化多数病例表达α-SMA、MSA或desmin,约50%病例表达ALK1,少数病例表达CK和CD68[15]。⑤淋巴结转移性癌:淋巴结结构多有部分残存,癌细胞免疫表型AE1/AE3、EMA等上皮标记阳性有助鉴别诊断。
3.4 治疗及预后 AFH宜采取局部广泛切除。几篇大宗报道文献数据显示,AFH局部复发率为13%左右(29/229),转移率约5%(12/229),一般转移到区域淋巴结。复发患者中包括多次局部复发者,再次手术多数仍然有效;偶因发生远处转移而导致死亡,病死率不到2%(4/229)[1-4]。局部复发可能与手术切除范围不够、肿瘤边界不规则及位于头颈部有关,局部或远处转移与瘤组织浸润到深筋膜或横纹肌有关[2]。本组2例均行局部肿块完整切除,镜下病变边界较清晰,无深部侵犯,切缘未见病变累及,随访54个月无复发及转移。
[1] Enzinger FM. Angiomatoid malignant fibrous histiocytoma: a distinct fibrohistiocytic tumor of children and young adults simulating a vascular neoplasm[J]. Cancer, 1979, 44(6):2147-2157.
[2] Costa MJ, Weiss SW. Angiomatoid malignant fibrous histiocytoma. A follow-up study of 108 cases with evaluation of possible histologic predictors of outcome[J]. Am J Surg Pathol, 1990, 14(12):1126-1132.
[3] Fanburg-Smith JC, Miettinen M. Angiomatoid “malignant” fibrous histiocytoma: a clinicopathologic study of 158 cases and further exploration of the myoid phenotype[J]. Am J Surg Pathol,1999, 30(11):1336-1343.
[4] 范钦和, Allen PW. 血管瘤样恶性纤维组织细胞瘤[J]. 中华病理学杂志, 1996, 25(1): 30-33.
[5] Fletcher CD, Bridge JA, Hogendoorn P,etal. WHO Classification of Tumours of Soft Tissue and Bone[M]. 4th edition. Lyon: IARC Press, 2013: 204-205.
[6] 王正, 范钦和, 王坚, 等. 实体型血管瘤样纤维组织细胞瘤临床病理观察[J]. 中华病理学杂志, 2013, 42(11):744-747.
[7] Chen G, Folpe AL, Colby TV,etal. Angiomatoid fibrous histiocytoma: unusual sites and unusual morphology[J]. Mod Pathol, 2011, 24(12):1560-1570.
[8] Bauer A, Jackson B, Marner E,etal. Angiomatoid fibroushistiocytoma: a case report and review of the literature[J]. J Radiol Case Rep, 2012, 6(11):8-15.
[9] Kao YC, Lan J, Tai HC,etal. Angiomatoid fibrous histiocytoma: clinicopathological and molecular characterisation with emphasis on variant histomorphology[J]. J Clin Pathol, 2014,67(3): 210-215.
[10] Thway K, Gonzalez D, Wren D,etal. Angiomatoid fibrous histiocytoma: comparison of fluorescence in situ hybridization and reverse transcription polymerase chain reaction as adjunct diagnostic modalities[J]. Ann Diagn Pathol, 2015, 19(3): 137-142.
[11] Bohman SL, Goldblum JR, Rubin BP,etal. Angiomatoid fibrous histiocytoma: an expansion of the clinical and histological spectrum[J]. Pathology, 2014, 46(3): 199-204.
[12] Tanas MR, Rubin BP, Montgomery EA,etal. Utility of FISH in the diagnosis of angiomatoid fibrous histiocytoma: a series of 18 cases[J]. Mod Pathol, 2010, 23(1):93-97.
[13] Thway K, Fisher C. Angiomatoid fibrous histiocytoma: the current status of pathology and genetics[J]. Arch Pathol Lab Med, 2015, 139(5): 674-682.
[14] 武忠弼, 杨光华. 中华病理学[M]. 北京: 人民卫生出版, 2002, 2426-2427.
[15] 王坚, 朱雄增. 软组织肿瘤病理学[M]. 北京: 人民卫生出版, 2008, 365-560.
Clinicopathologic features of angiomatoid fibrous histiocytoma
JIAO Nanlin, LU Linming, ZHANG Fan, WU Xiuxiu, XU Guoxiang
Department of Pathology, The first Affiliated Hospital of Wannan Medical College, Wuhu 241001, China
Objective:To understand the clinicopathological and genetic features of angiomatoid fibrous histiocytoma (AFH) for addressing the diagnosis and differential diagnosis of this entity.Methods:Clinicopahtological pictures of AFH were examined in 2 cases by histopathology, immunohistochemistry and fluorescence in situ hybridization (FISH), and the related literatures were reviewed.Results:The patients aged 9 and 24 years, and presented with a mass at elbow and neck, respectively. Both patients underwent simple excision. Gross examination showed a single mass with a clear boundary, measuring 1.5-2.0 cm in diameter. Microscopically, the distinctive histopathology were: ①aggregated nodules of spindle or histocytoid cells, one with significant atypia, and the mitotic figures were all below two per 10 HPF; ②peritumoral chronic inflammatory cells infiltration; ③tumor cells surrounded by a dense fibrous pseudocapsule; and ④blood-filled cystic spaces being seen in one cases. The margins were negative in both cases. Immunohistochemical staining showed that tumor cells were positive for vimentin, focally positive for CD68, CD99, rarely positive for SMA and EMA. MyoD1, Myogenin, Myoglobin, CD21, CD23, CD35, CD34, ALK-1, S-100 protein, HMB45 and CKpan were all negative. Proliferation index was 3%-10%. EWSR1 rearrangement was detected in both cases by FISH. Follow-up in 54 months showed no evidence of recurrence.Conclusions: AFH is an uncommon tumor and its correct diagnosis relies on clinical features and pathological examination. Immunohistochemical staining is useful for differential diagnosis, and EWSR1 rearrangement may be a potential biomarker for diagnosis.
fibrous histiocytoma, angiomatoid; immunohistochemistry; EWSR1
1002-0217(2015)05-0437-04
皖南医学院中青年科研基金项目(WK201035F)
2015-06-10
焦南林(1977-), 男, 主治医师, 硕士, (电话) 0553-5739792, (电子信箱) nanlinjiao@163.com; 张 帆, 男, 主任医师, (电子信箱) zhangfan401401@aliyun.com, 通讯作者.
R 738
A
