YPED:An Integrated Bioinformatics Suite and Database for Mass Spectrometry-based Proteomics Research
2015-02-06ChristopherColangeloMarkShifmanKeiHoiCheungKathrynStoneNicholasCarrieroErolGulcicekTuKietLamTerenceWu9RobertBjornsonCanBruce10AngusNairnJesseRinehartPerryMillerKennethWilliams
Christopher M.Colangelo*,Mark Shifman,Kei-Hoi Cheung, Kathryn L.StoneNicholas J.Carriero,Erol E.Gulcicek TuKiet T.LamTerence Wu9,Robert D.Bjornson,Can Bruce10,b, Angus C.Nairn,Jesse Rinehart,Perry L.Miller,6, Kenneth R.Williams
1W.M.Keck Foundation Biotechnology Resource Laboratory,School of Medicine,Yale University,New Haven,CT 06510,USA
2Department of Molecular Biophysics&Biochemistry,Yale University,New Haven,CT 06520,USA
3Yale Center for Medical Informatics,School of Medicine,Yale University,New Haven,CT 06510,USA
4Department of Anesthesiology,School of Medicine,Yale University,New Haven,CT 06510,USA
5Department of Emergency Medicine,School of Medicine,Yale University,New Haven,CT 06510,USA
6VA Connecticut Healthcare System,West Haven,CT 06516,USA
7Department of Computer Science,Yale University,New Haven,CT 06520,USA
8Yale Center for Genome Analysis,West Campus,Yale University,Orange,CT 06477,USA
9Yale West Campus Analytical Core,West Campus,Yale University,West Haven,CT 06516,USA
10Yale Bioinformatics Resource,School of Medicine,Yale University,New Haven,CT 06510,USA
11Department of Psychiatry,School of Medicine,Yale University,New Haven,CT 06510,USA
12Department of Cellular&Molecular Physiology,School of Medicine,Yale University,New Haven,CT 06510,USA
13Systems Biology Institute,Yale University,West Haven,CT 06516,USA
YPED:An Integrated Bioinformatics Suite and Database for Mass Spectrometry-based Proteomics Research
Christopher M.Colangelo1,2,*,a,Mark Shifman3,4,Kei-Hoi Cheung3,5,6, Kathryn L.Stone1,2,Nicholas J.Carriero1,7,8,Erol E.Gulcicek1,2, TuKiet T.Lam1,2,Terence Wu1,2,9,Robert D.Bjornson1,7,8,Can Bruce1,2,10,b, Angus C.Nairn11,c,Jesse Rinehart12,13,Perry L.Miller3,4,6, Kenneth R.Williams1,2
1W.M.Keck Foundation Biotechnology Resource Laboratory,School of Medicine,Yale University,New Haven,CT 06510,USA
2Department of Molecular Biophysics&Biochemistry,Yale University,New Haven,CT 06520,USA
3Yale Center for Medical Informatics,School of Medicine,Yale University,New Haven,CT 06510,USA
4Department of Anesthesiology,School of Medicine,Yale University,New Haven,CT 06510,USA
5Department of Emergency Medicine,School of Medicine,Yale University,New Haven,CT 06510,USA
6VA Connecticut Healthcare System,West Haven,CT 06516,USA
7Department of Computer Science,Yale University,New Haven,CT 06520,USA
8Yale Center for Genome Analysis,West Campus,Yale University,Orange,CT 06477,USA
9Yale West Campus Analytical Core,West Campus,Yale University,West Haven,CT 06516,USA
10Yale Bioinformatics Resource,School of Medicine,Yale University,New Haven,CT 06510,USA
11Department of Psychiatry,School of Medicine,Yale University,New Haven,CT 06510,USA
12Department of Cellular&Molecular Physiology,School of Medicine,Yale University,New Haven,CT 06510,USA
13Systems Biology Institute,Yale University,West Haven,CT 06516,USA
Proteomics;
We report a signifcantly-enhanced bioinformatics suite and database for proteomics research called Yale Protein Expression Database(YPED)that is used by investigators at more than 300 institutions worldwide.YPED meets the data management,archival,and analysis needs of a high-throughput mass spectrometry-based proteomics research ranging from a single
Mass spectrometry;
Repository;
Spectral library laboratory,group of laboratories within and beyond an institution,to the entire proteomics community.The current version is a signifcant improvement over the frst version in that it contains new modules for liquid chromatography–tandem mass spectrometry(LC–MS/MS)database search results,label and label-free quantitative proteomic analysis,and several scoring outputs for phosphopeptide site localization.In addition,we have added both peptide and protein comparative analysis tools to enable pairwise analysis of distinct peptides/proteins in each sample and of overlapping peptides/proteins between all samples in multiple datasets.We have also implemented a targeted proteomics module for automated multiple reaction monitoring(MRM)/selective reaction monitoring(SRM)assay development.We have linked YPED’s database search results and both label-based and label-free fold-change analysis to the Skyline Panorama repository for online spectra visualization.In addition,we have built enhanced functionality to curate peptide identifcations into an MS/MS peptide spectral library for all of our protein database search identifcation results.
Introduction
Proteomics is a key method for advancing our understanding of biological processes and systems.Making this technology accessible to the biological community is critically important [1].The rapid growth of mass spectrometry(MS)data in proteomics research has necessitated the creation of new bioinformatics tools and databases to effciently pull together diverse sets of analyses.With the growing use of high-throughput proteomics technologies in life science research,there is a call for‘‘democratizing’’proteomics data[2],that is,making the source data in scientifc publications available to the readers. Although making MS data publicly available has still not been widely mandated by journals as a requirement for publication, a number of public databases have been created for accepting data submissions(post-publication or as part of the publication process)from the proteomics community.As reviewed by Vizcaı´no et al.[3],these databases include the Global Proteome Machine(GPM)[4],Proteomics Identifcations database(PRIDE)[5],and PeptideAtlas[6].The 2014 NAR database registry provides a more comprehensive list of public proteomics resources(http://www.oxfordjournals.org/nar/database/cat/10)such as Model Organism Protein Expression Database(MOPED)[7]and Plasma Proteome Database (PPD)[8].
While the importance of sharing proteomics data broadly has been emphasized[9],the kind and format of data and metadata to share(in addition to when to share the data)have been an active topic of discussion in the proteomics community.This has led to a number of proteomics data standard initiatives(http://www.psidev.info)[10].One of these initiatives is the Minimum Inofrmation about A Proteomics Experiment (MIAPE)[11],whose goal is to specify the information necessary to interpret the results of the proteomics experiment unambiguously and to potentially reproduce the results of the experiment.
As the amount of public proteomics data increases rapidly, concerns have been raised regarding data quality.For example,Schaab et al.[12]have pointed out the issue of data quality existing in public proteomics databases due to heterogeneous sources.This makes data comparison and integration diffcult across proteomics experiments conducted independently by different research groups.To address issues such as these,we developed Yale Protein Expression Database(YPED;version 1.0)[13]as a uniform system for collecting proteomic data derived from multiple samples that have been submitted by hundreds of investigators for analysis in the Keck Foundation Biotechnology Resource Laboratory at Yale University.This uniformity of sample entry into YPED ensures that only precise and high quality data,e.g.,protein identifcation results fltered with 1%false discovery rate (FDR),are curated for future proteomic experimentation. Subsequently,other laboratories have implemented data fltering models such as MaxQB[12]and Panorama[14](http://proteome.gs.washington.edu/software/skyline).In addition to discovery proteomics,targeted proteomic assays have become more common[15].Therefore,there is a growing need for proteome data to be well curated into MS/MS spectral libraries and formore integrative multiple reaction monitoring (MRM)/selective reaction monitoring(SRM)tools to be developed [15].Several public libraries already exist,such as PeptideAtlas[6],SRMAtlas[16],NationalInstitute of Standards and Technology(NIST)Libraries of Peptide Tandem Mass Spectra(http://peptide.nist.gov/),GPMDB[4], and the PeptideAtlas SRM Experiment Library(PASSEL) [17].However,these libraries often require expert user intervention to generate MRM/SRM transition lists.

Figure 1 Workfow diagram summarizing YPED system components and their relationships
In light of these challenges,we present here a signifcantlyenhanced version of YPED,an open-source proteomics suite and database[13].Figure 1displays the main components ofthe YPED system.In contrast to laboratory-specifc and community-based proteomics databases,YPED is unique in providing a comprehensive workfow that extends from sample submission through a web user interface,which provides immediate access to newly-acquired data,to an integrated suite of biostatistical and bioinformatics tools for analyzing the resulting mass spectrometric proteomics data.On the other hand,YPED consists of both a local database and a public repository that provides access to published and anonymous results.The wide range of data access privileges of YPED enables it to meet the needs of individual,multiple collaborative,and core laboratories.It supports multiple MS instruments and search engines.It also supports quantitation of labeled and label-free proteomics data.Sample/project annotations and search results stored in the database can be queried and viewed via a web user interface.We have also developed and integrated a suite of statistical analysis tools to enhance the quality and visualization of data.In addition,the YPED system is interoperable with a number of external resources to leverage proteomics databases and tools created by other groups.The source code of the YPED system can be downloaded from http://yped.med.yale.edu/yped_dist/. A demo accountwith Usernameasyped_demo and Password as yped_demo contains representative data results.
YPED’s increasingly important role in biomedical research is highlighted by its usage statistics.As of January 12,2015, YPED contained 18,985 datasets from 1654 users in the laboratories of 702 principal investigators at more than 300 institutions around the world.These datasets contained liquid chromatography(LC)-MS/MS analyses from 3,997,386 distinct peptides derived from 929,665 distinct proteins. YPED’s spectral library contains spectra from 340,449 distinct human peptides,which are more than the 293,000 non-redundant spectra used by Kim et al.[18]to map the human proteome.YPED’s spectral library contains≥2 distinct peptides from 19,327 human,16,154 mouse,7661 rat,6007 yeast,and 4080Escherichia coliproteins,respectively.
Methods
User statistics and summary
YPED is a web-accessible,password-protected database for managing high-throughput proteomic analyses.For a comprehensive,current usage statistics report for YPED that is updated daily please visit:https://yped.med.yale.edu:8443/ yp_results/QDSTATS_report.do.We have extended YPED’s functionality to keep in step with rapidly-evolving MS and proteomic technologies.The initial report(YPED version 1.0)[13]described analysis requisition,result reporting and sample comparison for multi-dimensional protein identifcation technology(MudPIT)[19],difference gel electrophoresis (DIGE)[20],and isotope-coded affnity tag(ICAT)labeled [21]samples.In addition,YPED now includes modules for LC–MS peptide and protein identifcations(LC–MS/MS), multiplexed isobaric tagging technology(iTRAQ[22]and tandem mass tag(TMT)[23]),stable isotope labeling by amino acids in cell culture(SILAC)[24],LC–MS/MS label-free quantitation[25](Skyline and Progenesis),and scoring for phosphopeptide localization(Mascot Delta Score(MD-score) [26]and PhosphoRS[27]).Using the discovery proteomic results,we have built a MRM/SRM targeted proteomics pipeline that includes an MS/MS spectral library.The peptide sequences in the spectral library have been compared via protein BLAST[28]against Swiss-Prot and TrEMBL databases [29]to determine if these sequences are unique to a specifc protein and organism.
Individual researchers can access their data through a simple user interface(Figure 2).Principal investigators(PIs)can also access all datasets generated by staff from within their laboratories.Individual experimental results are listed as samples,which can then be grouped into projects to help researchers keep track of different stages of their project.Each sample contains the experimental felds necessary to meet the MIAPE sample guidelines,including information such as sample preparation protocols,proteomics instrumentation and methodology,so results can be reproduced and compared.Not only does this data organization/annotation enhance data sharing,but it also facilitates the publication process.A publication can be associated with one or multiple samples and/ or projects.Researchers can view,subset and download their data through the secure web interface.There are also proteomics core‘‘superuser’’accounts(Figure S1)that allow multiple staff in one or more proteomics cores to upload MS data. In addition,YPED also features modules for sample submission,tracking,and billing.The‘‘regular’’user interface (Figure 2)contains three sections:the project listings,sample listings,and user functions such as search,sample requisition, and project management. The ‘‘superuser’’ interface (Figure S1)provides the ability to carry out many additional options such as sample submission,project management,sample tracking,data import,sample administration,and user billing.Additionally,within projects,superusers or users can organize and provide additional documentation to their datasets by linking raw data and/or associated documents(e.g., PDF and PowerPoint fles).
System implementation
YPED is available as an open-source package.The web application is written in Java using Struts(version 1.3.10).The web server is confgured using Tomcat 7.0.20 and connects to an Oracle database (version 11g).Italso connectsto a Windows-based fle server through fle transfer protocol (FTP).The source code,javadoc and oracle schema can all be downloaded from the web page(http://yped.med.yale. edu/yped_dist/).
Results
LC–MS protein identifcation

Figure 2 YPED PI/User main menu
Version 1.0 of YPED supported ProteinProphet(protXML) and PeptideProphet(pepXML).In the extended version we added an LC–MS module to include results from Mascot (Matrix Science Inc.)search(current version 2.4.0)and ProteinPilot.Mascot results are imported after transformation into an XML fle employing the Mascot script, export_dat_2.pl. YPED also supports ProteinPilot (Paragon)*.group result fles that have been converted to an XML document.We then developed an XML schema definition(ProteinPilot4.xsd)that enables either of the resulting XML fles to be parsed and loaded into YPED using JAXB (http://jaxb.java.net/) and Java StAX API (http://stax. codehaus.org/).These results can be viewed via the web and include FDRs,the proteins identifed with scores and coverage maps,and peptides identifed for each protein with attendant peptide scores(Figure 3).Data are presented via a browser in tables where summary facts can be conveniently browsed using hyperlinks,enabling users to drill all the way down to the MS/MS data.Users have the option of additionally processing their protein identifcation data through ProteinProphet(protXML)and PeptideProphet(pepXML) and displaying the combined results(Figure S2).YPED also contains additional protein identifcation information such as the exponentially-modifed protein abundance index(emPAI) [30],which enables estimation of absolute protein amount within a complex proteome sample.Although the emPAI results are not displayed on the main LC result page,they are contained in the exported Excel spreadsheet(Figure S3).
Label-based quantitative analysis
iTRAQ[22]and TMT reagents[31]allow multiplexing of protein samples and produce identical MS spectra but label specifc reporter fragment ions for the multiple versions of the labeled peptide.YPED currently supports mass spectrometric data processing with either ProteinPilot[32](AB Sciex Inc.)or Mascot software.Both packages perform protein identifcation and peptide reporter ion quantitation.Protein and peptidedata resultsfrom ProteinPilotare exported as comma-delimited text fles(.csv format)and imported into YPED.For Mascot iTRAQ/TMT quantitation results,both the protein identifcations and peptide reporter ions are imported as described in the above LC–MS protein identifcation section(Figure S4).
SILAC[24]studies can be processed by initial database searching with Mascot and then using the quantitation toolbox in Mascot Distiller(Matrix Science Inc.).The resulting Mascot distiller XML output is then processed with JAXB and the Java StAX API before insertion into YPED.The web results page displays the LC–MS results along with the heavy/light ratios and SILAC peptides.
Label-free quantitative analysis

Figure 3 YPED LC–MS result page
LC–MS/MS label-free quantitation data can be processed with either Skyline or Progenesis LC–MS software(NonlinearDynamics,LLC),with Skyline also enabling analysis of LC–SWATH datasets.For Skyline,the peak integration results are uploaded to Panorama and also exported to a comma delimited text(*.csv)fle.The text fle is then uploaded to YPED,where these results are merged to generate a report table as shown for SWATH data inFigure 4.This report contains protein ID,peptide sequence,isotope dot product,and quantitation values.In addition,YPED contains links to the stored chromatograms on Panorama,where users can visualize their Skyline peak integration results(Figure 4).Label-free Progenesis LC–MS results are exported to Excel,parsed with the POI Java library(http://poi.apache.org/),and inserted into YPED.The Mascot search results are imported as described above.YPED merges both these results to generate a web report table(Figure 5)that contains protein ID,confdence scores,quantitation values,ratios and ANOVAPvalues with options for generating a Volcano plot of the results.In addition,individual peptide identifcations can be conveniently browsed using hyperlinks,enabling users to drill all the way down to the MS/MS data.
Phosphoprotein analysis
To leverage newly-developed tools that help to identify sites of peptide phosphorylation,YPED was upgraded to include both phosphoprotein flters and phosphopeptide scoring algorithms to aid in site localization analysis.These upgrades enable researchers to automate phosphopeptide site localization on large LC–MS datasets and have high confdence that the site assignments are correct.To access the phosphoprotein flter from the LC–MS,SILAC,or label-free quantitation results, users simply click the hyperlink,‘‘View PhosphoProteins’’, which then brings up a web page that displays a listing of the phosphoproteins identifed and the number of phosphopeptide matches for each protein.Further navigation can be done by clicking the‘‘view’’hyperlink under the phosphopeptide column in the table,after which YPED will then generate a table containing rows of identifed phosphopeptides with each phosphorylated amino acid underlined and with columns containing the associated MD-score[26],PhosphoRS[27] probability score,m/z,ion mass,mass accuracy(ppm),and peptide charge(Figure 6).
Comparative analysis
Tools have been added to facilitate downstream sample comparison and to assess the distribution of biological functions (through a remote query to PANTHER[33])among the identifed proteins in a sample.For downstream analysis,researchers can compare samples based on peptide or protein content, or cross-compare the proteins from various analyses such as comparing a MudPIT to an iTRAQ analysis.A pairwise analysis on each sample is performed and the results are listed in a table format with distinct peptides/proteins in each sample and the peptides that overlap between all samples(Figure S5).
Targeted proteomics
An entire targeted proteomics pipeline has been integrated into YPED,which enables utilization of our custom peptide spectral library database(see below)to facilitate peptide and MRM/SRM transition selection for global targeted proteomic analysis,tools for method export,and an interface for collation of quantitation data results and review.Specifcally, transitions and retention times can be rapidly retrieved from database search results to guide the validation of complex large-scale discovery studies by MRM-based targeted proteomics.To generate a targeted proteomics experiment,users frst query the entire YPED spectral library using the‘‘Protein ID Peptide Report’’search tool,which has flters forprotein accession numbers,protein names,peptide sequences,and gene symbols.YPED then displays the search results in a browser,where users select peptides to add to a targeted proteomics experiment list.When the list is fnalized, YPED automatically flters proteins/peptides on the server without the need for expert user intervention,thereby maximizing productivity.YPED uses the following criteria for fltering. First,peptide scores have to be greater than or equal to the identity score.Second,proteins must have three or more peptides.Third,peptides that match 1 protein in the given species specifc BLASTP[28]search are kept.Fourth,peptides containing methionine residues are excluded.Finally,the remaining peptides are sorted based on their number of occurrences in YPED with the top peptides being chosen for downstream MRM/SRM analysis.After peptide selection,the highest ion intensities are selected as transitions for downstream MRM/ SRM analysis.These MRM/SRM transitions along with their retention times are exported as a tab-delimited fle(tsv)and then used to populate a targeted mass spectrometer method fle.
Spectral library for downstream MRM/SRM assay development
The spectral library is generated by frst taking each Mascot search result and fltering it at 1%FDR.Then all the unique LC–MS peptide identifcations with Mascot peptide scores greater than homology and 5–30 amino acids in length are compared to the Swiss-Prot database using a protein BLAST search[28].Table 1shows a summary of the BLASTP results for fve model organisms commonly used in proteomic analyses.The BLASTP results are stored in YPED as a table which includes the number of observations per peptide and each individual observation.After BLAST analysis,we fltered the number of proteins to 19,327 for human,16,154 for mouse, and 7661 for rat with two or more distinct peptides per protein. These results are then used to verify that a given set of candidate peptides are unique to a protein when determining targeted(SRM/MRM)candidates for future assays.We also have implemented the ability to export either individual samples or a project(series of samples)from Mascot search fles to BiblioSpec format[34]utilizing Blibbuild.The resulting spectral libraries can be utilized in searching MS/MS spectra [35]or for Skyline.
Public repository
We have developed a publicly-accessible YPED repository to further increase accessibility to YPED’s proteomics data (http://yped.med.yale.edu/repository)(Figure 7).It contains the results of projects that have been released for public viewing by the principal investigators along with raw data from the samples.To broaden the visibility and interoperability,we have also released the project results to the NeuroscienceInformation Framework(NIF)federated data repository (https://www.neuinfo.org/mynif/databaseList.php).This allows YPED to be integrated with a wide variety of neuroscience databases to enhance its support of neuroproteomics research.

Figure 4 Skyline Label-free SWATH results in YPED
The YPED repository also has an access code provision for viewing results prior to public release.This feature is useful for making the results available to reviewers and collaborators who do not have YPED access.

Figure 5 Screenshot of the Label-free quantitation data results
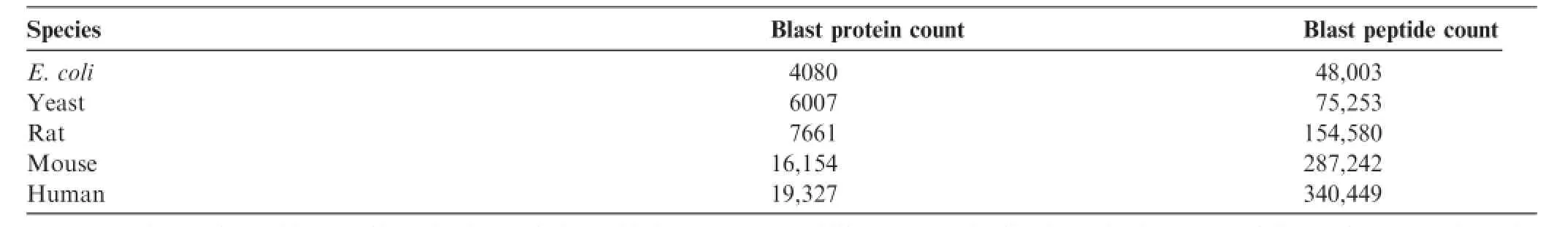
Table 1 YPED spectral library BLAST results(UniProtKB/SwissProt Database)
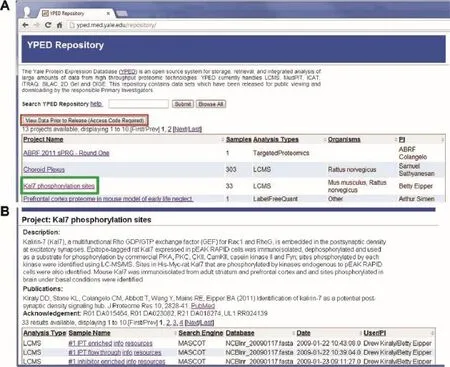
Figure 7 YPED Repository
The repository provides a query interface to search anonymous results based on protein IDs/names,peptide sequence and gene symbols.Figure S6 shows a portion of the search results for a protein whose ID is KCC2G_HUMAN.The search returns 51 distinct peptides above the peptide score threshold.
Discussion
To tackle the huge data challenges posed by high-throughput LC/MS/MS proteomics datasets,we have assembled a team from a broad range of disciplines including benchscientists,clinicians,computer scientists(with database and high-performance computing expertise),bioinformaticians, biostatisticians,and proteomics technologists.Such a multidisciplinary approach was a key to developing YPED into a userfriendly,scalable,evolvable and sustainable resource.The resulting YPED is an integrated suite of tools designed to cover a broad spectrum of techniques for quantitative proteomics(discovery and targeted proteomics;and labeled and label-free quantitation).It captures data produced by a wide range of MS instruments and technologies,and presents them via the web as a set of relevant results that are understandable for non-specialists.
YPED implements a wide range of data access privileges associated with different user types including core laboratory users,researchers(PIs and their laboratory members),and public users.One advantage of this approach is that it allows data sharing at different levels.For example,researchers can share their data within a specifc laboratory and/or between laboratories(possibly located at different institutions).Core facility users can help individual laboratories to populate data in YPED as they have read/write access to the laboratories they work with.YPED was started with one core facility (Keck Foundation Biotechnology Resource Laboratory at Yale).Recently,we have added another core proteomics facility,West Campus Analytical Chemistry Core that is part of the West Campus expansion at Yale.In the future,we may be able to add core facilities beyond Yale who are willing to adhere to the same high standards of data quality(e.g.,1%FDR fltered protein identifcation results).In addition to security,the different user roles facilitate collaboration in a trusted environment.
The frst version of YPED[13]only supported a few technologies,but as mass spectrometric methods have evolved we extended YPED(version 2.0)to handle these new data types.Ongoing work includes integrating YPED to handle additionalquantitative techniques and programs (e.g., Maxquant and data-independent analysis such as SWATH [36])and to update as new instruments are obtained.We also would welcome the opportunity to expand YPED’s linkage to externaldatabases/knowledgebasessuchasPRIDE or PeptideAtlas.In addition to PANTHER,we will enable YPED to incorporate information from pathway and protein network resources such as KEGG[37],Reactome[38],and STRING[39].
While we will continue to address the needs of individual laboratories,we also will increase our interaction with the proteomics community,such as the Association of Biomolecular Resource Facilities (ABRF; http://www.abrf.org/) and Human Proteome Organization(HUPO;http://www.hupo. org/),to help promote the use and development of standards (e.g.,HUPO-PSI[40])for exchanging data with other major proteomics databases(e.g.,PRIDE,GPM,PASSEL and PeptideAtlas).For example,in addition to producing our spectral libraries in BiblioSpec format,we are working to support mzIdentML (http://www.psidev.info/mzidentml), since a growing number of tools support these standardized formats. As biomedical ontologies have increasingly been applied to proteomics databases such as PRIDE,we will also explore the use of ontologies to standardize proteomic data annotation and enable ontologically-based data integration.Finally,we have created a virtual machine for YPED that greatly increases the fexibility and ease of future deployment of YPED to other institutions or into a shared infrastructure(e.g.,in the cloud) accessed by multiple institutions.
Author’s contributions
KHC,PLM and KRW supervised the study.CMC oversaw the design of YPED and MS wrote the source code for YPED.CMC wrote the manuscript and ACN edited the manuscript.KLS,NJC,EEC,TTL,TW,RDB,CB,and JR all contributed ideas for improving YPED.All authors read and approved the fnal manuscript.
Competing interests
The authors declared that there are no competing interests.
Acknowledgments
We would like to thank Hans Aerni for comments and review of the manuscript.This project was supported in part by the National Institutes of Health of the United States(Grant Nos.UL1 RR024139 to Yale Clinical and Translational Science Award,1S10OD018034-01 to 6500 QTrap Mass Spectrometer for Yale University,1S10RR026707-01 to 5500 QTrap Mass Spectrometer for Yale University,P30 DA018343toYale/NIDANeuroproteomicsCenterand NIDDK-K01DK089006 awarded to JR).
Supplementary material
Supplementary material associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j. gpb.2014.11.002.
[1]Kenyon G,DeMarini D,Fuchs E,Galas DJ,Kirsch J,Leyh T, et al.Defning the mandate of proteomics in the post-genomics era:workshop report.Mol Cell Proteomics 2002;10:763–80.
[2]Editorial.Democratizing proteomics data.NatBiotechnol 2007;25:26.
[3]Vizcaı´no J,Foster J,Martens L.Proteomics data repositories: providing a safe haven for your data and acting as a springboard for further research.J Proteomics 2010;73:2136–46.
[4]Craig R,Cortens J,Beavis R.Open source system for analyzing, validating,and storing protein identifcation data.J Proteome Res 2004;3:1234–42.
[5]Martens L,Hermjakob H,Jones P,Adamski M,Taylor C,States D,et al.PRIDE:the proteomicsidentifcationsdatabase. Proteomics 2005;5:3537–45.
[6]Deutsch W,Lam H,Aebersold R.PeptideAtlas:a resource for target selection for emerging targeted proteomics workfows. EMBO Rep 2008;9:429–34.
[7]Kolker E,Higdon R,Haynes W,Welch D,Broomall W,Lancet D,et al.MOPED:ModelOrganism Protein Expression Database.Nucleic Acids Res 2012;40:D1093–9.
[8]NanjappaV,ThomasJK,MarimuthuA,MuthusamyB, Radhakrishnan A,Sharma R,et al.Plasma Proteome Database as a resource for proteomics research:2014 update.Nucleic Acids Res 2014;42:D959–65.
[9]Editorial.Thou shalt share your data.Nat Methods 2008;5:209.
[10]Orchard S,Hermjakob H,Julian R,Runte K,Sherman D,Wojcik J,et al.Common interchange standards for proteomics data: public availability of tools and schema.Proteomics 2004;4:490–1.
[11]Taylor CF,Paton NW,Lilley KS,Binz PA,Julian RK,Jones AR, et al.The minimum information about a proteomics experiment (MIAPE).Nat Biotechnol 2007;25:887–93.
[12]Schaab C,Geiger T,Stoehr G,Cox J,Mann M.Analysis of high accuracy,quantitative proteomics data in the MaxQB database. Mol Cell Proteomics 2012;11,M111.014068.
[13]Shifman M,Li Y,Colangelo C,KS KL,Wu T,Cheung K,et al. YPED:a web-accessible database system for protein expression analysis.J Proteome Res 2007;6:4019–24.
[14]Sharma V,Eckels J,Taylor GK,Shulman NJ,Stergachis AB, Joyner SA,et al.Panorama:a targeted proteomics knowledge base.J Proteome Res 2014;13:4205–10.
[15]Huttenhain R,Malmstrom J,Picotti P,Aebersold R.Perspectives of targeted mass spectrometry for protein biomarker verifcation. Curr Opin Chem Biol 2009;13:518–25.
[16]Picotti P,Lam H,Campbell D,Deutsch E,Mirzaei H,Ranish J, et al.A database of mass spectrometric assays for the yeast proteome.Nat Methods 2008;5:913–4.
[17]Farrah T,Deutsch E,Kreisberg R,Sun Z,Campbell D,Mendoza L,et al.PASSEL:the PeptideAtlas SRMexperiment library. Proteomics 2012;12:1170–5.
[18]Kim MS,Pinto SM,Getnet D,Nirujogi RS,Manda SS, Chaerkady R,et al.A draft map of the human proteome. Nature 2014;509:575–81.
[19]Washburn MP,Wolters D,Yates 3rd JR.Large-scale analysis of the yeast proteome by multidimensional protein identifcation technology.Nat Biotechnol 2001;19:242–7.
[20]Unlu¨M,Morgan M,Minden J.Difference gel electrophoresis:a single gel method for detecting changes in protein extracts. Electrophoresis 1997;18:2071–7.
[21]Gygi SP,Rist B,Gerber SA,Turecek F,Gelb MH,Aebersold R. Quantitative analysis of complex protein mixtures using isotopecoded affnity tags.Nat Biotechnol 1999;17:994–9.
[22]Ross P,Huang Y,Marchese J,Williamson B,Parker K,Hattan S, et al.Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents.Mol Cell Proteomics 2004;3:1154–69.
[23]Thompson A,Schafer J,Kuhn K,Kienle S,Schwarz J,Schmidt G,et al.Tandem mass tags:a novel quantifcation strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 2003;75:1895–904.
[24]Ong S,Blagoev B,Kratchmarova I,Kristensen D,Steen H, Pandey A,et al.Stable isotope labeling by amino acids in cell culture,SILAC,as a simple and accurate approach to expression proteomics.Mol Cell Proteomics 2002;1:376–8.
[25]Asara J,Christofk H,Freimark L,Cantley L.A label-free quantifcation method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics 2008;8:994–9.
[26]Savitsk M,Lemeer S,Boesche M,Lang M,Mathieson T, Bantscheff M,et al.Confdent phosphorylation site localization using the Mascot Delta Score.Mol Cell Proteomics 2011;10, M110.003830.
[27]Taus T,Ko¨cher T,Pichler P,Paschke C,Schmidt A,Henrich C, et al.Universal and confdent phosphorylation site localization using phosphoRS.J Proteome Res 2011;10:5354–62.
[28]Altschul SF,Gish W,Miller W,Myers EW,Lipman DJ.Basic local alignment search tool.J Mol Biol 1990;215:403–10.
[29]Bairoch A,Boeckmann B.The SWISS-PROT protein sequence data bank:current status.Nucleic Acids Res 1994;22:3578–80.
[30]Ishihama Y,Oda Y,Tabata T,Sato T,Nagasu T,Rappsilber J, et al.Exponentially modifed protein abundance index(emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein.Mol Cell Proteomics 2005;4:1265–72.
[31]Shilov I,Seymour S,Patel A,Loboda A,Tang W,Keating S, et al.The Paragon Algorithm,a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra.Mol Cell Proteomics 2007;6:1638–55.
[32]Cui C,Churchchill G.Statistical tests for differential expression in cDNA microarray experiments.Genome Biol 2003;4:210.
[33]Mi H,Muruganujan A,Thomas PD.PANTHER in 2013: modeling the evolution of gene function,and other gene attributes,in the context of phylogenetic trees.Nucleic Acids Res 2013;41:D377–86.
[34]Frewen BE,Merrihew GE,Wu CC,Noble WS,MacCoss MJ. Analysis of peptide MS/MS spectra from large-scale proteomics experiments using spectrum libraries. Anal Chem 2006;78:5678–84.
[35]Frewen B,MacCoss M.Using BiblioSpec for creating and searching tandem MS peptide libraries. Curr Protoc Bioinformatics 2007;13,Unit 13.7.
[36]Gillet L,Navarro P,Tate S,Ro¨st H,Selevsek N,Reiter L,et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition:a new concept for consistent and accurateproteomeanalysis.MolCellProteomics2012;11, O111.016717.
[37]Kanehisa M,Goto S.KEGG:Kyoto Encyclopedia of Genes and Genomes.Nucleic Acids Res 2000;28:27–30.
[38]Joshi-Tope G,Gillespie M,Vastrik I,D’Eustachio P,Schmidt E, de Bono B,et al.Reactome:a knowledgebase of biological pathways.Nucleic Acids Res 2005;33:D428–32.
[39]Szklarczyk D,Franceschini A,Kuhn M,Simonovic M,Roth A, Minguez P,et al.The STRING database in 2011:functional interaction networks of proteins,globally integrated and scored. Nucleic Acids Res 2011;39:D561–8.
[40]Orchard S,Binz P,Borchers C,Gilson M,Jones A,Nicola G, et al.Ten years of standardizing proteomic data:a report on the HUPO-PSI Spring Workshop:April 12–14th,2012,San Diego USA.Proteomics 2012;12:2767–72.
Received 25 July 2014;revised 13 October 2014;accepted 11 November 2014
Available online 21 February 2015
Handled by Xiaowen Liu
*Corresponding author.
E-mail:christopher.colangelo@yale.edu(Colangelo CM).
aORCID:0000-0002-0438-1395.
bORCID:0000-0002-3101-1250.
cORCID:0000-0002-7075-0195.
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
http://dx.doi.org/10.1016/j.gpb.2014.11.002
1672-0229©2015 The Authors.Production and hosting by Elsevier B.V.on behalf of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY license(http://creativecommons.org/licenses/by/4.0/).
Database;
Bioinformatics;
