A Brief Review of Software Tools for Pangenomics
2015-02-06JingfaXiaoZhewenZhangJiayanWuJunYu
Jingfa Xiao*,Zhewen Zhang,Jiayan Wu,Jun Yu
CAS Key Laboratory of Genome Sciences and Information,Beijing Institute of Genomics,Chinese Academy of Sciences, Beijing 100101,China
A Brief Review of Software Tools for Pangenomics
Jingfa Xiao*,a,Zhewen Zhangb,Jiayan Wuc,Jun Yud
CAS Key Laboratory of Genome Sciences and Information,Beijing Institute of Genomics,Chinese Academy of Sciences, Beijing 100101,China
Pangenomics;
Pangenome;
Comparative analysis;
Genomic dynamics;
Core genes
Since the proposal for pangenomic study,there have been a dozen software tools actively in use for pangenomic analysis.By the end of 2014,Panseq and the pan-genomes analysis pipeline (PGAP)ranked as the top two most popular packages according to cumulative citations of peerreviewed scientifc publications.The functions of the software packages and tools,albeit variable among them,include categorizing orthologous genes,calculating pangenomic profles,integrating gene annotations,and constructing phylogenies.As epigenomic elements are being gradually revealed in prokaryotes,it is expected that pangenomic databases and toolkits have to be extended to handle information of detailed functional annotations for genes and non-protein-coding sequences including non-coding RNAs,insertion elements,and conserved structural elements.To develop better bioinformatic tools,user feedback and integration of novel features are both of essence.
Introduction
In the past decade or so,the remarkable advancement of DNA sequencing technology and application has led to an astronomical accumulation of genomic data.This is especially true for the prokaryotic genomes as individual of them is only a few megabases in size.It is expected that in the next decade or two, there will be more data collected than what we can actually handle.Therefore,database construction,improvement,and
*Corresponding author.
E-mail:xiaojf@big.ac.cn(Xiao J).
aORCID:0000-0002-2835-4340.
bORCID:0000-0002-9422-822X.
cORCID:0000-0001-6048-405X.
dORCID:0000-0002-2702-055X.
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China. consolidation,as well as new tool development,are especially welcome.In this way,the sibling felds of genomics,such as pangenomics and metagenomics,can all be ready for curating, sharing,and mining foods of the incoming genomic big data. Coming back to the reality and focusing on pangenomics, there were,as of December 2014,more than 40 bacterial species that have over 20 fully-assembled genomes from different strains and isolates,allowing for comprehensive pangenomic studies.The concept of pangenome was frst proposed in 2005 by Tettelin et al.[1,2],which is defned as the entire genomic repertoire of a given species or phylogenetic clade when multiple species are defned by systematics.According to the defnition,gene profle(content)of a pangenome is divided into three groups:core(shared by all genomes),dispensable, and strain-(or isolate-)specifc genes.A series of pangenomic studies have been performed in genomic dynamics[3–6], pathogenesis and drug resistance[7–9],bacterial toxins[10],and species evolution[11].The concept has also been extended to viral[12],plant[13–15],and fungal genome studies[16].A review on ten-year history and feld achievement of pangenomics has just been published at the beginning of 2014[2], which detailed major projects as well as methodology and technology advancements.
Here,we provide a brief review on the pangenomic software packages and tools,including their basic function,general utility,and popularity based on their cumulative citation by peer-reviewed scientifc publications.Although such a singlecriterion evaluation may never be adequate and thorough,we hope that it provides a feld guide for students and young scientists to make the right choice for their preferred applications.
Highlights of the software packages and tools
Since 2010,we have seen a dozen or so software packages and tools being put forward,which are capable of clustering orthologous genes,identifying single nucleotide polymorphisms(SNPs),constructing phylogenies,and profling core/ shared/isolate-specifc genes.Although they may share similar functions,each has its own characteristics and limitations, leaving rooms for further improvement.
Among the early-developed packages,Panseq[17]and PanCGHweb[18]were published in 2010,followed by CAMBer[19]and the Prokaryotic-genome Analysis Tool (PGAT)[20]in 2011.PanCGHweb is a web tool for pangenomic microarray analysis based on PanCGH algorithm[21]. It enables users to group genes into orthologs and to construct gene-based phylogenies ofrelated strains and isolates. However,this package is rather specifc for handling microarray data but not RNA-seq data.Panseq,another online pangenomic tool,is able to determine core and accessory regions of genome assemblies based on MUMmer and BLASTn,as well as to identify SNPs among the core genomic regions.In addition,Panseq also has a locus selector module that selects the most discriminatory loci among the accessory loci or core gene SNPs[17].Panseq,however,is not able to provide pangenomic profle and functional enrichment analysis that is important for the biologists to flter out functional relevance of the pangenomic elements.The later released CAMBer is designed to identify multi-gene families from multiple bacterial strains and isolates.These multi-gene families can be used for sequencing errordetection,mutation identifcation,and pangenomic profle computation[19].CAMBer is supreme in refning gene function prediction according to multi-gene family information,but it does not provide tools for comparative or evolutionary analysis among strains and isolates.As a webbased database,PGAT integrates several useful functions, such as plotting the presence and absence of genes among members of a pangenome,identifying SNPs among orthologs and syntenic regions,comparing gene orders among different strains and isolates,providing KEGG pathway analysis tools, and searching for genes through different annotations such as the Cluster of Orthologous Groups of proteins(COG), PSORT,SignalP,the Tied Mixture Hidden Markov Model (TMHMM),and Pfam.However,PGAT is just a database with a limited number of species curated and it cannot perform analysis for new sequencing data from users.
PGAP is a stand-alone program developed by Zhao et al.in 2012,which contains fve functional models[22].Based on functional gene clustering and analysis,PGAP presents pangenomic profle(partitions of pangenomic elements or gene categories),genetic variation,species evolution,and function enrichment of different strains and isolates of a given pangenome.In addition,all analyses are performed with a single command,and such integration is rather user-friendly and effcient. Nonetheless,PGAP has its limitation as well.For instance,all its output fles of the fve models are text fles,which lacks of intuitiveness.Contreras-Moreira et al.subsequently proposed a program called GET_HOMOLOGUES in 2013,which is also a versatile software package for pangenomics[23].This software package integrated data download,sequence feature extraction,homologous gene identifcation,pangenome profling,graphical display,and phylogenetic tree construction into one powerful toolkit.Several other tools were also available in 2013,such as PanCake[24]and PANNOTATOR[25]. PanCake was developed for identifying singletons and core regions in arbitrary sequence sets,while PANNOTATOR,a web-based automated pipeline,was designed for the annotation of closely-related genomes for pangenomic analysis.However, these two tools only focus on simple functions,such as clustering homologous genes and gene curation.In 2014,a powerful and fexible toolkit,the Integrated Toolkit for Exploration of microbial Pan-genomes(ITEP),was published by Benedict and colleagues[26].ITEP integrates plenty of existing bioinformatics tools for pangenomic analysis,including protein family prediction,ortholog detection,functional domain analysis, pangenomic profling,and metabolic network integration. Moreover,ITEP also integrates some visualization scripts that assist biologists in phylogenetic tree construction,annotation curation,and specifc query for conserved protein domain identifcation.In 2014,another rapid core-genome alignment and visualization pangenomic software package,Harvest,was proposed by Treangen et al.Harvest contains tools,such as Parsnp and Gingr,for core gene alignment,variant calling,recombination detection,and phylogenetic trees construction[27].To analyzepangenomic profle in a larger scale,a software package PanGP was developed with a graphic interface by Zhao et al.in 2014[28].Spine and AGEnt were also developed in 2014,which are capable of profling pangenomes based on both fnished and draft genomic sequences[29].We summarized all the software packages and tools inTable 1,highlighting their platforms and major features.We went one step further and ranked them according to their citations by peer-reviewed scientifc publications(Figure 1),which were collected from ISI Web of Science-Science Citation Index Expanded.Our summary indicates that Panseq and PGAP have been the most popular packages up to the end of 2014.
A wish list for improving the current software
Although single-tool solution could not usually satisfy the need for understanding the whole picture,a wish list from the users is always helpful for prioritizing goals for the package developers,hence providing directions for improving each package.
First,the performance of pangenomic analysis strongly depends on the accuracy of genome assembly and annotation. Therefore,an adequate number of complete sequence assemblies are a prerequisite.Currently,most of the existing bacterial genome sequences are actually incomplete(in most of thecases,contigs are not joined together into single chromosomes),and some only have high-quality and high-coverage raw data available.The inclusion of incomplete genome assemblies for pangenomic analysis may need scaffold building that requires reformatting of the contig data fles.Despite the development of the third-generation sequencing technology,which would certainly help the assembly and fnishing of prokaryotic genomes[30],incomplete prokaryotic genomes are expected to be deposited into public databases in mass.It would be a waste if such data are left unused.

Table 1 Software tools for pangenomic studies
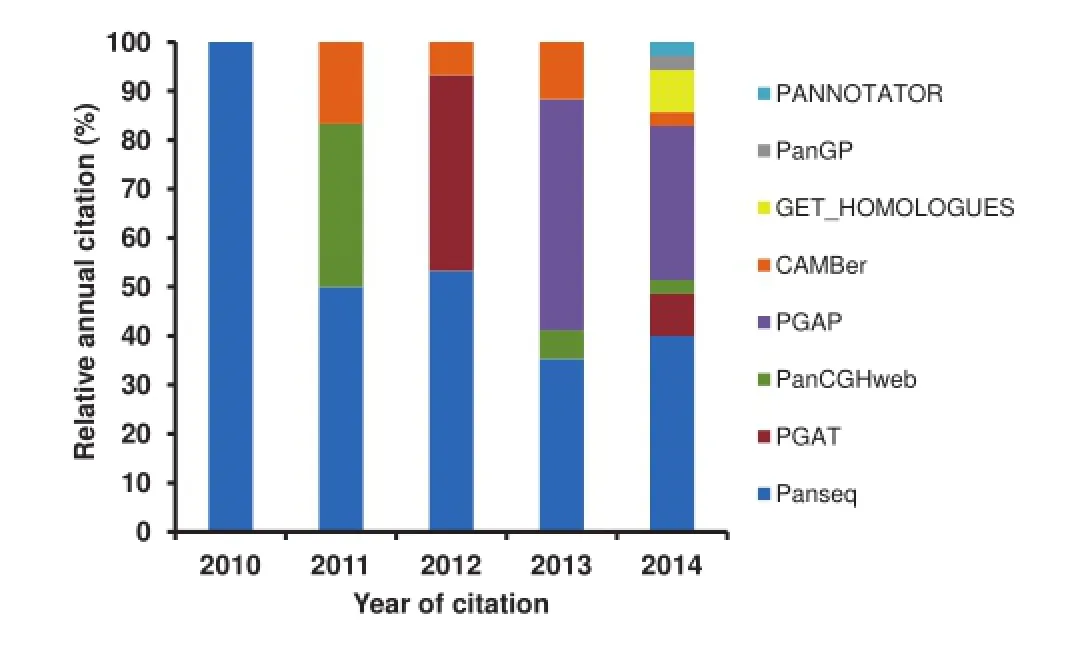
Figure 1 Relative citation of the pangenomic software tools from peer-reviewed scientifc publications
Second,orthologous gene identifcation is a key step in pangenomic analysis.At present,the existing software for ortholog detection is mainly based on sequence similarity, phylogenetic relationship,or other annotation information such as functional information.The development of novel and more effcient ortholog identifcation method for multiple closely-related strains and isolates can greatly improve the accuracy of pangenomic analysis.One possibility is to integrate gene gain-and-loss information for phylogeny building among strains and isolates.
Third,sampling is also important for pangenomics in a couple of counts.One is how many strains or isolates to choose for a pangenomic analysis.The other is how to implement a flter that differentiates more diverse strains or isolates from the less diverse for pangenomic analysis.For instance,if we choose all genomes of a species for an analysis,which include one or a few divergent genomes,the core genome will be much shorter or reduced.Obviously,individual genomes should be selected and regrouped for better representation of average nucleotide identity(ANI).ANI is one of the most useful measurements for species delineation[31].Therefore,for a better pangenomic analysis,detailed information for the available samples is of essence,which should include their genotypes, phenotypes,and habitats.
Fourth,the current tools have not incorporated some recent advancements in prokaryotic genomics,such as the so-called genome-organization frameworks(GOFs),which are not only unique to each species but also provide guidance for sequence assembly and fnishing[32].Other annotation information,such as that of non-coding RNAs,pseudogenes, and epigenetic elements,remains to be implemented into the relevantsoftwarepackages.Finally,a never-ending improvementofpangenomictoolsisvisualization that provides not only better displays but also quality graphics for publication.
Concluding remarks
We provide an overview on the existing pangenomic analysis tools and hope to see improvements of the software tools from their original developers.We certainly express our enthusiasm for new tools to join the competition,and after all,for a piece of bioinformatic work,a database or a toolkit,the survival or winning game is in its long-term maintenance and constant improvement.
Competing interests
The authors declared that there are no competing interests.
Acknowledgements
This study was supported by the National High-tech R&D Program(863 Program;Grant No.2012AA020409)from theMinistry of Science and Technology of China,the Key Program of the Chinese Academy of Sciences(Grant No. KSZD-EW-TZ-009-02),and the National Natural Science Foundation of China(Grant Nos.31471248 and 31271386).
[1]Tettelin H,Masignani V,Cieslewicz MJ,Donati C,Medini D, Ward NL,et al.Genome analysis of multiple pathogenic isolates ofStreptococcus agalactiae:implications for the microbial‘‘pangenome’’.Proc Natl Acad Sci U S A 2005;102:13950–5.
[2]Vernikos G,Medini D,Riley DR,Tettelin H.Ten years of pangenome analyses.Curr Opin Microbiol 2014;23C:148–54.
[3]Rasmussen TB,Danielsen M,Valina O,Garrigues C,Johansen E, Pedersen MB.Streptococcus thermophiluscore genome:comparative genome hybridization study of 47 strains.Appl Environ Microbiol 2008;74:4703–10.
[4]WillenbrockH,HallinPF,WassenaarTM,UsseryDW. Characterization of probioticEscherichia coliisolates with a novel pan-genome microarray.Genome Biol 2007;8:R267.
[5]Zhang A,Yang M,Hu P,Wu J,Chen B,Hua Y,et al. Comparative genomic analysis ofStreptococcus suisreveals signifcant genomic diversity among different serotypes.BMC Genomics 2011;12:523.
[6]KaasRS,FriisC,UsseryDW,AarestrupFM.Estimatingvariation within the genes and inferring the phylogeny of 186 sequenced diverseEscherichia coligenomes.BMC Genomics 2012;13:577.
[7]Park J,Zhang Y,Buboltz AM,Zhang X,Schuster SC,Ahuja U, et al.Comparative genomics of the classicalBordetellasubspecies: the evolution and exchange of virulence-associated diversity amongst closely related pathogens.BMC Genomics 2012;13:545.
[8]D’Auria G,Jimenez-Hernandez N,Peris-Bondia F,Moya A, Latorre A.Legionella pneumophilapangenome reveals strainspecifc virulence factors.BMC Genomics 2010;11:181.
[9]Hu P,Yang M,Zhang A,Wu J,Chen B,Hua Y,et al. Comparative genomics study of multi-drug-resistance mechanisms in the antibiotic-resistantStreptococcus suisR61 strain.PLoS One 2011;6:e24988.
[10]Fang Y,Li Z,Liu J,Shu C,Wang X,Zhang X,et al.A pangenomic study ofBacillus thuringiensis.J Genet Genomics 2011;38:567–76.
[11]Konstantinidis KT,Ramette A,Tiedje JM.The bacterial species defnition in the genomic era.Philos Trans R Soc Lond B Biol Sci 2006;361:1929–40.
[12]AherfS,Pagnier I,Fournous G,Raoult D,La Scola B,Colson P. CompletegenomesequenceofCannes8virus,anewmemberofthe proposed family‘‘Marseilleviridae’’.Virus Genes 2013;47:550–5.
[13]Cao J,Schneeberger K,Ossowski S,Gunther T,Bender S,Fitz J, et al.Whole-genome sequencing of multipleArabidopsis thalianapopulations.Nat Genet 2011;43:956–63.
[14]Read BA,Kegel J,Klute MJ,Kuo A,Lefebvre SC,Maumus F, et al.Pan genome of the phytoplanktonEmilianiaunderpins its global distribution.Nature 2013;499:209–13.
[15]Li YH,Zhou G,Ma J,Jiang W,Jin LG,Zhang Z,et al.De novoassembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits.Nat Biotechnol 2014;32:1045–52.
[16]Dunn B,Richter C,Kvitek DJ,Pugh T,Sherlock G.Analysis of theSaccharomyces cerevisiaepan-genome reveals a pool of copy number variants distributed in diverse yeast strains from differing industrial environments.Genome Res 2012;22:908–24.
[17]Laing C,Buchanan C,Taboada EN,Zhang YX,Kropinski A, Villegas A,et al.Pan-genome sequence analysis using Panseq:an online tool for the rapid analysis of core and accessory genomic regions.BMC Bioinformatics 2010;11:461.
[18]Bayjanov JR,Siezen RJ,van Hijum SAFT.PanCGHweb:a web tool for genotype calling in pangenome CGH data.Bioinformatics 2010;26:1256–7.
[19]Wozniak M,Wong L,Tiuryn J.CAMBer:an approach to support comparative analysis of multiple bacterial strains.BMC Genomics 2011;12:S6.
[20]Brittnacher MJ,Fong C,Hayden HS,Jacobs MA,Radey M, Rohmer L.PGAT:a multistrain analysis resource for microbial genomes.Bioinformatics 2011;27:2429–30.
[21]Bayjanov JR,Wels M,Starrenburg M,van Hylckama Vlieg JE, Siezen RJ,Molenaar D.PanCGH:a genotype-calling algorithm for pangenome CGH data.Bioinformatics 2009;25:309–14.
[22]Zhao Y,Wu J,Yang J,Sun S,Xiao J,Yu J.PGAP:pan-genomes analysis pipeline.Bioinformatics 2012;28:416–8.
[23]Contreras-Moreira B,Vinuesa P.GET_HOMOLOGUES,a versatile software package for scalable and robust microbial pangenome analysis.Appl Environ Microbiol 2013;79:7696–701.
[24]Ernst C,Rahmann S.PanCake:a data structure for pangenomes. In:Beißbarth T,Kollmar M,Leha A,Morgenstern B,Schultz A, editors.German conference on bioinformatics 2013.Dagstuhl, Germany:Schloss Dagstuhl-Leibniz-Zentrum fuer Informatik; 2013.p.35–45.http://dx.doi.org/10.4230/OASIcs.GCB.2013.35.
[25]Santos AR,Barbosa E,Fiaux K,Zurita-Turk M,Chaitankar V, Kamapantula B,et al.PANNOTATOR:an automated tool for annotation of pan-genomes.Genet Mol Res 2013;12:2982–9.
[26]Benedict MN,Henriksen JR,Metcalf WW,Whitaker RJ,Price ND.ITEP:an integrated toolkit for exploration of microbial pangenomes.BMC Genomics 2014;15:8.
[27]Treangen TJ,Ondov BD,Koren S,Phillippy AM.The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecifc microbial genomes.Genome Biol 2014;15:524.
[28]Zhao Y,Jia X,Yang J,Ling Y,Zhang Z,Yu J,et al.PanGP:a toolfor quickly analyzing bacterialpan-genome profle. Bioinformatics 2014;30:1297–9.
[29]Ozer EA,Allen JP,Hauser AR.Characterization of the core and accessory genomes ofPseudomonas aeruginosausing bioinformatic tools Spine and AGEnt.BMC Genomics 2014;15:737.
[30]Koren S,Harhay GP,Smith TP,Bono JL,Harhay DM,Mcvey SD,et al.Reducing assembly complexity of microbial genomes with single-molecule sequencing.Genome Biol 2013;14:R101.
[31]Kim M,Oh HS,Park SC,Chun J.Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol 2014;64:346–51.
[32]Kang Y,Gu C,Yuan L,Wang Y,Zhu Y,Li X,et al.Flexibility and symmetry of prokaryotic genome rearrangement reveal lineage-associated core-gene-defned genome organizational frameworks.MBio 2014;5:e01867.
Received 9 January 2015;revised 22 January 2015;accepted 25 January 2015
Available online 23 February 2015
Handled by Vladimir Bajic
http://dx.doi.org/10.1016/j.gpb.2015.01.007
1672-0229©2015 The Authors.Production and hosting by Elsevier B.V.on behalf of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
