礞石滚痰丸中金胺O与金橙Ⅱ的检测方法
2015-01-16吴嫣艳于倩茜
吴嫣艳, 于倩茜
(1.江苏省食品药品监督检验研究院,江苏南京210008;2.南京市江宁中医院,江苏南京211100)
[质量]
礞石滚痰丸中金胺O与金橙Ⅱ的检测方法
吴嫣艳1, 于倩茜2
(1.江苏省食品药品监督检验研究院,江苏南京210008;2.南京市江宁中医院,江苏南京211100)
目的建立检测礞石滚痰丸中2种染色剂 (金胺O与金橙Ⅱ)的方法。方法礞石滚痰丸70%乙醇提取,采用液质联用,高效液相色谱柱为Agela Venusil MP C18(4.6 mm×250 mm,5μm),流动相A为0.02 mol/L乙酸铵溶液,流动相B为乙腈,梯度洗脱,检测波长为430 nm(金胺O)和485 nm(金橙Ⅱ)。结果HPLC法金胺O在2.084~41.680μg/mL范围内、金橙Ⅱ在2.292~45.840μg/mL范围内线性关系良好,金胺O、金橙Ⅱ检测限分别为1.202、0.444 ng,平均回收率为90.9%、94.3%;LC/MS/MS法金胺O、金橙Ⅱ检测限分别为0.305、0.430 ng。结论本方法快速灵敏可靠,可用于礞石滚痰丸中金胺O与金橙Ⅱ的检测。
礞石滚痰丸;液质联用;金胺O;金橙Ⅱ;非法添加
礞石滚痰丸为2013年国家评价性抽验品种,收载于 《中国药典》2010年版一部,处方中含有金礞石、沉香、黄芩、熟大黄四味中药。具有逐痰降火的功效,用于痰火扰心所致的癫狂惊悸,或喘咳痰稠、大便秘结。笔者针对安全性方面做了探索性研究。
近年来,用劣质黄芩非法染色以改善外观,提高售价现象在市场上屡见不鲜。考虑到礞石滚痰丸处方中有黄芩,本实验参考有关文献采用HPLC、LC/MS/MS法建立了礞石滚痰丸中可能非法添加的2种染色剂 (金胺O、金橙Ⅱ) 的检测方法[1-16]。本方法快速、准确、灵敏、可靠,可用于礞石滚痰丸中可能非法添加金胺O、金橙Ⅱ的检测。
1仪器与试剂试药
1.1 仪器 Agilent 1260 HPLC;SHIMADZU LC-10AD HPLC;Agilent1100 Series LC/MSD Trap。
1.2 材料试剂 14批礞石滚痰丸为2013年国家评价性抽验样品,由江苏省食品药品监督检验研究院提供;金胺O对照品 (批号111770-201302)与金橙Ⅱ对照品 (批号111769-200701)均来自中国食品药品检定研究院。
乙腈,色谱纯,Fisher Scientific公司;乙酸铵,色谱纯,Sigma公司;水为超纯水;其余试剂为分析纯。
2 方法与结果
2.1 高效液相色谱法
2.1.1 色谱条件与系统适应性试验 Agela Venusil MP C18色谱柱 (4.6 mm×250 mm,5μm);流动相A为0.02 mol/L乙酸铵溶液,流动相B为乙腈,梯度洗脱(0~10 min,A相65%;10~11 min,A相65%→50%;11~18 min,A相50%;18~19 min,A相50%→75%;19~25 min,A相75%;25~27 min,A相75%→20%;27~36 min,A相20%;36~38 min,A相20%→65%;38~60 min,A相 65%);体积流量为 1.0 mL/min;检测波长为430 nm(金胺O)、485 nm(金橙Ⅱ);柱温为35℃;进样量为10μL。各成分的理论塔板数均>50 000,相邻峰的分离度均>1.5。对照品色谱图及样品典型色谱图见图1、图2。
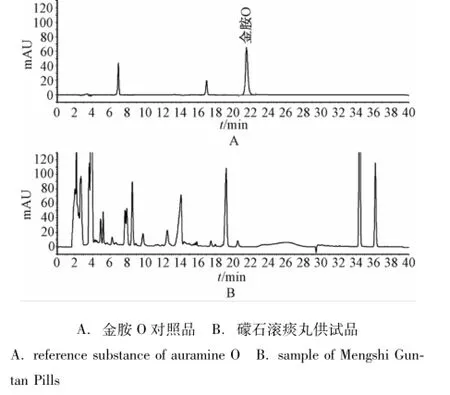
图1 检测波长430 nm下的高效液相色谱图Fig.1 HPLC chromatogram s by 430 nm
2.1.2 对照品溶液的制备 取金胺O和金橙Ⅱ对照品适量,精密称定,加70%乙醇制成质量浓度各为20μg/mL的混合对照品溶液,即得。
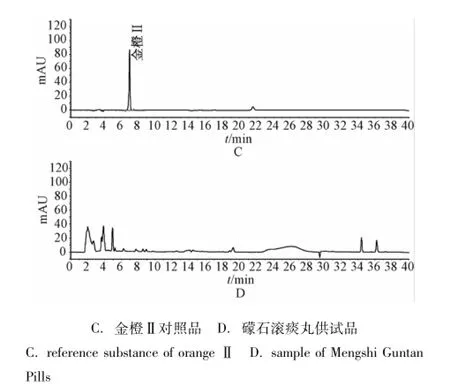
图2 检测波长485 nm下的高效液相色谱图Fig.2 HPLC chrom atogram s by 485 nm
2.1.3 供试品溶液的制备 取礞石滚痰丸适量,研细,取约1 g,精密称定,加70%乙醇10 mL,超声 (功率为135 W,频率为42 kHz)提取20 min,12 000 r/min离心10 min,取上清液为供试品溶液。
2.1.4 线性关系考察 精密称取金胺O、金橙Ⅱ对照品适量,加70%乙醇制成质量浓度分别为2、4、10、20、30、40μg/mL的系列对照品溶液,在“2.1.1”项所述色谱条件下分别进样10μL。以质量浓度 (μg/mL)为横坐标,峰面积为纵坐标,绘制标准曲线,结果见表1。

表1 线性关系考察数据Tab.1 Data of linear relation test
2.1.5 精密度试验 取混合对照品溶液 (20 μg/mL),在“2.1.1”项所述色谱条件下连续进样6次,记录峰面积。结果金胺O的RSD为0.32%、金橙Ⅱ的RSD为0.19%,表明仪器精密度良好。
2.1.6 加样回收试验 取礞石滚痰丸样品适量,研细,取约1 g,精密称定,精密加入质量浓度均为200μg/m L的金胺O、金橙Ⅱ混合对照品溶液1 mL,精密加入70%乙醇9 mL,超声 (功率为135 W,频率为42 kHz)提取20 min,12 000 r/min离心10min,取上清液,即得。测定并计算。结果平均回收率(n=6)金胺O为90.9%(RSD= 1.5%)、金橙Ⅱ为94.3%(RSD=1.2%)。
2.1.7 稳定性试验 取同一份加样回收供试品溶液分别于0、2、4、8、12、16、24 h进行测定,结果金胺O的RSD为0.83%、金橙Ⅱ的RSD为0.52%,表明溶液在24 h内基本稳定。
2.1.8 HPLC检测限 取“2.1.4”项下最低浓度对照品溶液加70%乙醇逐步稀释,按 “2.1.1”项所述色谱条件进样,信噪比大于3时,金胺O的检测限为1.202 ng、金橙Ⅱ的检测限为0.444 ng。
2.1.9 样品测定 取礞石滚痰丸样品按上述方法测定,结果14批样品在金胺O、金橙Ⅱ对照品保留时间处均未检出相应的色谱峰。
2.2 液质联用法
2.2.1 条件与参数
2.2.1.1 色谱条件 色谱柱采用Waters Xbridge Shield RP18柱(4.6 mm×250 mm,5μm);流动相A为0.02 mol/L乙酸铵溶液,流动相B为乙腈,梯度洗脱(0~10 min,A相65%;10~11 min,A相65%→50%;11~18 min,A相50%;18~20 min,A相50%→20%;20~30 min,A相20%;30~31 min,A相20%→65%;31~45 min,A相65%);体积流量为1.0 mL/min;检测波长为430 nm(金胺O)、485 nm(金橙Ⅱ);柱温为35℃;进样量为10μL。分流进样,分流比为4∶1。
2.2.1.2 质谱参数 ESI正负离子检测模式(正离子模式检测金胺O,负离子模式检测金橙Ⅱ),雾化器35.0 psi(1 psi=6.895 kPa),干燥气体积流量9.0 L/min,干燥气温度350℃,正离子裂解电压0.80 V,负离子裂解电压1.50 V,扫描范围50~700 m/z。
2.2.2 对照品溶液的制备 即 “2.1.2”项下对照品溶液。
2.2.3 供试品溶液的制备 即 “2.1.3”项下礞石滚痰丸供试品溶液。
2.2.4 测定结果 取对照品溶液和供试品溶液,按 “2.2.1”项下条件与参数进样,记录质谱图。结果14批礞石滚痰丸均未检出与金胺O、金橙Ⅱ对照品一致的质谱峰。质谱图见图3、图4。
2.2.5 LC/MS/MS检测限 将质谱对照品溶液逐渐稀释,用以确定方法的检出限 (S/N>3),金胺O检测限为0.305 ng,金橙Ⅱ检测限为0.430 ng。
3 讨论
液质联用法试验时分别采用正离子与负离子方式检测,发现金胺O、金橙Ⅱ分别是正离子方式、负离子方式检测灵敏、稳定,故最终选择金胺O采用正离子方式检测,金橙Ⅱ采用负离子方式检测。
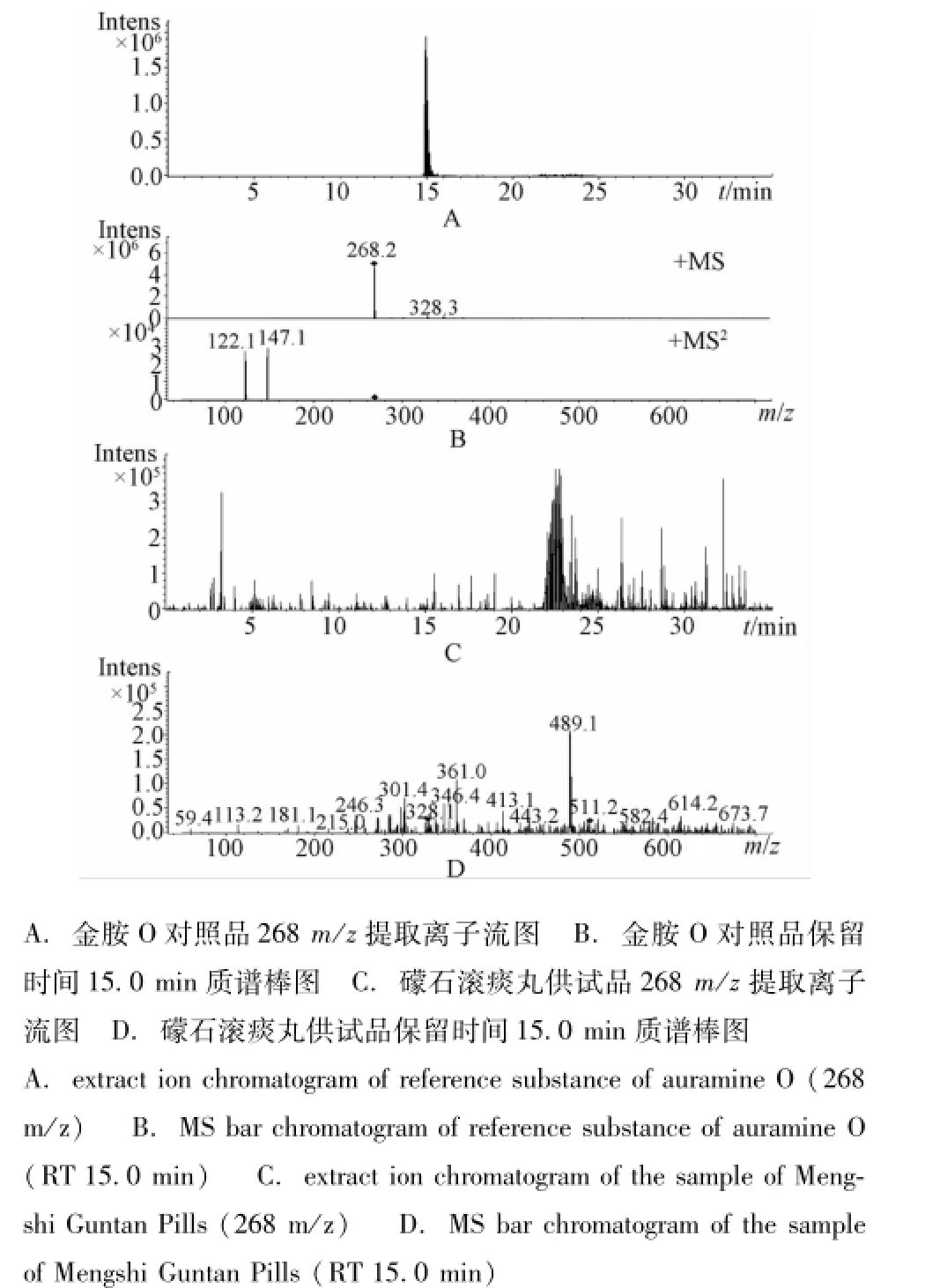
图3 质谱图ESI+Fig.3 LC/MS/MS chrom atogram s by ESI+
中药材及中药饮片是中成药质量的源头,而目前染色药材和饮片在市场上常有发现,势必存在影响中成药质量的可能。为规范市场,保障人民群众安全用药,针对中成药中非法添加染色剂的检测方法研究十分必要。
本研究采用高效液相色谱法和液质联用技术,对礞石滚痰丸中是否含有金胺O、金橙Ⅱ进行检测,建立的方法快速简便准确可靠,可为礞石滚痰丸的质量控制提供参考。
[1]葛会奇,贾天柱.LC-MS/PAD法测定非法染色的中药蒲黄和黄芩饮片中金胺 O[J].辽宁中医药杂志,2011,38(8):1616-1618.
[2]蒋 玲,彭飞燕,饶伟文,等.染色黄连中金胺O的检测方法研究[J].中草药,2011,42(7):1344-1347.
[3]肖 凌,侯俊杰,聂 晶,等.元胡止痛系列制剂中金胺O的检测[J].药物分析杂志,2012,32(11):2008-2011.

图4 质谱图ESIFig.4 LC/MS/MS chromatogram s by ESI-
[4]蒋 磊,孟祥松,姬凤娟.HPLC-DAD法检测女宝胶囊中的微量金橙Ⅱ[J].安徽医药,2011,15(4):437-439.
[5]梁选革,张若燕,刘莉丽.HPLC法测定红花注射液中色素金橙Ⅱ[J].中国药事,2012,26(2):137-139.
[6]高天兵.中药材专业市场部分中药材质量状况研究[J].中国药事,2009,23(2):155-156.
[7]郑 娟,邹耀华.HPLC-PDA法检测蒲黄和黄连中十种非法添加色素[J].中国卫生检验杂志,2011,21(5):1078-1082.
[8]胡晓炜.超高效液相色谱法检测西红花中非法添加的染料金橙Ⅱ[J].药物鉴定,2011,20(14):40-41.
[9]蒋永梅.丹参中非法添加金橙Ⅱ的检测[J].西北药学杂志,2010,25(4):245-246.
[10]莫连峰,饶伟文,赵纯玉.高效液相色谱法测定染色黄芩中金胺的含量[J].药物鉴定,2007,16(15):29-30.
[11]石思佳,宋桂英,张 继.中药饮片的染色鉴别[J].北京中医药,2011,30(9):698-699.
[12]金 卓,丁 晴.骨折挫伤胶囊中非法染色剂金橙Ⅱ的检测方法研究[J].海峡药学,2012,24(7):64-67.
[13]饶伟文,蒋 玲,赵纯玉,等.几种染色掺假中药的化工染料鉴定[J].药物分析杂志,2007,27(11):1742-1745.
[14]富小珺,张春梅.几种染色药材中化工染料金胺O的定性鉴别[J].中国医药指南,2013,11(21):107-108.
[15]赵纯玉,饶伟文,莫连峰.蒲黄中金胺O的HPLC测定[J].药物分析杂志,2007,27(12):1956-1958.
[16]王卓晅.田七痛经胶囊中掺入色素金胺O的检查[J].药学与临床研究,2011,19(2):191-192.
Identification of auram ine O and orangeⅡin M engshi Guntan Pills
WU Yan-yan1, YU Qian-qian2
(1.Jiangsu Institute for Food and Drug Control,Nanjing 210008,China;2.Nanjing Jiangning Hospital of Traditional Chinese Medicine,Nanjing 211100,China)
AIMTo establish a method for detecting two coloring agents(auramine O and orangeⅡ)in Mengshi Guntan Pills.METHODSThe determination of70%ethanolic extractofMengshiGuntan Pillswasperformed on HPLC and LC/MS/MSmethod.Their column was Agela VenusilMPC18(4.6 mm×250 mm,5μm)with 0.02 mol/L ammonium acctate as mobile phase A and acetonitrile as mobile phase B in a gradient elution manner.The wavelength detection was430 nm for auramine O and 485 nm for orangeⅡ.RESULTSAuramine O and orangeⅡin HPLC chromatogram showed good linearity in the range of 2.084-41.680μg/mL and 2.292-45.840μg/mL,their detection's limits of auramine O and orangeⅡwere 1.202 ng and 0.444 ng,their average recoveries were 90.9%and 94.3%,respectively.The detection's limits of auramine O and orangeⅡin LC/MS/MSwere 0.305 ng and 0.430 ng,respectively.CONCLUSIONThemethod is rapid,sensitive and reliable,and it can be used for the determination ofMengshi Guntan Pillswith adulteration-auramine O and orangeⅡ.
:MengshiGuntan Pills;LC/MS/MS;auramine O;orangeⅡ;adulteration
R927.2
:A
:1001-1528(2015)06-1232-04
10.3969/j.issn.1001-1528.2015.06.016
2014-07-03
吴嫣艳 (1984—),女,主管药师,硕士,从事中药质量研究。Tel:(025)86632807,E-mail:21724604@qq.com
