Efects of Carbide Formation in Graphene Growth
2015-01-13ZhunzhunWangQiquanLuoWenhuaZhangZhenyuLi
Zhun-zhun WangQi-quan LuoWen-hua ZhangZhen-yu Li
Hefei National Laboratory for Physical Sciences at the Microscale,University of Science and Technology of China,Hefei 230026,China
Efects of Carbide Formation in Graphene Growth
Zhun-zhun Wang,Qi-quan Luo,Wen-hua Zhang,Zhen-yu Li∗
Hefei National Laboratory for Physical Sciences at the Microscale,University of Science and Technology of China,Hefei 230026,China
Besides carbon solubility,the carbide formation possibility is another important factor to diferentiate various substrate materials in graphene growth.A recent experiment indicates that the formation of transition metal carbides(TMCs)can suppress carbon precipitation. In this study,Mo2C,a representative of TMCs,is used to study the efects of carbide formation in graphene growth from frst principles.Carbon difusion in Mo2C bulk turns out to be very difcult and it becomes much easier on the Mo2C(001)surface.Therefore, carbon precipitation suppression and graphene growth can be realized simultaneously.A direction depended difusion behavior is observed on the Mo2C(101)surface,which makes it less favorable for graphene growth compared to the(001)surface.
Molybdenum carbide,Difusion,Density functional theory
I.INTRODUCTION
Due to its unique properties and potential applications[1,2],graphene has been intensively studied in the last decade.Among the several reported methods to synthesize graphene[3],chemical vapor deposition (CVD)is especially promising to obtain high quality samples at a large scale[4].To optimize a CVD growth, the substrate should be carefully chosen[5].Various metal substrates have been tried in graphene growth, and high quality monolayer graphene can be grown on some of them,such as Cu[6].
On diferent substrates,the graphene growth mechanisms can be very diferent[7−9].To choose a suitable metal substrate,we can check metal-C phase diagrams and compare corresponding parameters including carbon solubility and carbide formation possibility [10].Efects of the former have been intensively studied already[11].On metal substrates with a relatively high carbon solubility,graphene growth proceeds via a precipitation mechanism,which makes a layer number control difcult to achieve[12].In contrast,on metal substrates with negligible C solubility,such as Cu,graphene growth is limited on the surface.As a result,uniform graphene monolayer can be readily obtained[6].
The possibility of carbide formation is less studied in the context of graphene growth.Recently,Zouet al.[13]have successfully grown uniform monolayer graphene on early transition metals.It is suggested that the realization of a layer number control in their experiment critically depends on the carbide formation, which suppresses the upward segregation or precipitation of carbon.To fully understand the efects of carbide formation in graphene growth,it is desirable to study the atomic details of relevant processes such as carbon difusion in carbide.
In this study,using Mo as an example,efects of carbide formation in graphene growth are studied from frst principles.According to Mo-C phase diagram[14],the stable carbide phase at a typical growth temperature (1050◦C)is Mo2C.Carbon difusion in Mo2C bulk and on its surfaces is studied.Based on our calculations, a clear picture of the molybdenum carbide formation aided layer number control in graphene growth is obtained.
II.COMPUTATIONAL DETAILS
All calculations were performed with the density functional theory(DFT)implemented in the Viennaab initiosimulation package(VASP)[15,16]within the projector augmented wave(PAW)framework[17], using the Perdew-Burke-Ernzerhof(PBE)exchangecorrelation functional[18].A 450 eV kinetic energy cutof was chosen for the plane wave basis set[19].Geometry optimizations were done until forces were smaller than 0.02 eV/˚A and the energy diference was lower than 10−5eV.Monkhorst-Packk-point sampling[20] was used in our calculations with a careful convergence test.The climbing image nudged elastic band(CI-NEB) method[21]was used to locate transition states,where the residual forces were within 0.03 eV/˚A.In surface calculations,slab models were constructed with vacuum zones thicker than 20˚A.
To compare the stability of diferent types of defects, their chemical environment should be specifed,which



whereEtotis the energy of the whole system andEpristineis the energy of Mo2C bulk or surface without defect.The positive and negative signs correspond to carbon vacancy and doped/adsorbed carbon,respectively.
III.RESULTS AND DISCUSSION
A.Carbon difusion in Mo2C bulk
There are mainly two Mo2C crystalline structures,i.e.the orthorhombic[22]and hexagonal[23,24] phases.Following previous studies[25,26],we focus on the hexagonal phase,where Mo atoms form a hexagonally close packed structure and carbon atoms fll half of the octahedral interstitial sites(Fig.1).Each interstitial site has six neighboring Mo atom.There are two types of C and Mo atoms,forming diferent layers perpendicular to thecaxis.The calculated lattice parameters area=2×3.04˚A,b=2×3.04˚A,andc=4.72˚A,in good agreement with experiment values[27].Distance between two octahedral sites in theabplane is 2.63˚A, and along thecdirection it is 2.36˚A.
Since only half of the octahedral interstitial sites are occupied by C,a natural C difusion pathway in Mo2C bulk is from an occupied to a neighboring unoccupied octahedral site.C difusion can also proceed via defects,such as C vacancy and C interstitial,in Mo2C bulk.Before discussing these difusion pathways,we frst check the thermodynamic stabilities of diferent types of defects.Since it brings no obvious structure deformation,a 2×1×2 supercell is used to study carbon vacancy.However,if an extra C atom is doped in the Mo2C bulk,a structure relaxation around the dopant atom is found.Therefore,a larger 2×2×2 supercell is built to ensure that any two neighboring doping carbon atoms are separated more than 12˚A away and thus basically interaction free.As shown in Fig.2,in the whole range ofµC,formation energy of C vacancy is positive while that of interstitial C dopant is negative.Therefore,C vacancy is thermodynamically unfavorable even in C-poor conditions(low C chemical potential),while C atom doping can be easily realized especially in C-rich conditions(high C chemical potential).
For intrinsic C difusion from an occupied C site to a neighboring unoccupied octahedral site,there are two possible difusion directions according to the hexagonal symmetry.We name the occupied C site as siteα,the neighboring octahedral site along thecaxis as siteβand that within theabplane as siteγ(Fig.1(c)).In pristine bulk phase,these two unoccupied sites are equivalent. However,ifαsite carbon is removed,symmetry between sitesβandγis broken in our model.From siteαto siteβ,the difusion barrier is 2.65 eV and the fnal state is 1.17 eV higher in energy than the initial state.From siteαto siteγ,the difusion barrier is 3.56 eV and the fnal state becomes 2.00 eV higher in energy.
For a C dopant at siteβ,difusion to a neighboring octahedral site within theabplane has an energy barrier of 2.59 eV.The initial and fnal states are equivalent. From siteβ,a C dopant can also difuse to a farther octahedral site(siteγin Fig.1(c)).However,there isan intermediate state in the difusion path,as shown in Fig.3.The intermediate state is 1.77 eV higher in energy,where the C dopant is located at a tetrahedral site. To difuse fromβtoγ,two successive energy barriers of 2.39 and 1.47 eV should be conquered.

FIG.1(a)Top view and(b)side view of Mo2C unit cell. (c)A 2×1×2 supercell with some atomic sites marked.

FIG.2 Formation energy of carbon adatom/dopant and vacancy in Mo2C bulk and surface systems.
Although it is thermodynamically not very favorable, we have also studied vacancy mediated difusion for completeness.Considering a C atom at siteα,there are two kinds of neighboring vacancy sites which can help C difusion∶sitesδandεas shown in Fig.1(c). Theαδandαεdistances are 3.03 and 3.84˚A,respectively.If there is a C vacancy at siteδ,carbon difusion fromαtoδneeds to conquer a barrier of 3.81 eV.The difusion barrier for theαtoεpath is 2.77 eV.There is no energy diference between the initial and fnal states in vacancy mediated C difusion.
Therefore,carbon difusion in Mo2C bulk is always associated with a very high energy barrier,which will thus be difcult even at a graphene growth temperature (typically about 1300 K).As a result,the formation of molybdenum carbide can efectively suppress carbon segregation/precipitation,as indicated in the experiment[13].Notice that the consistency among diferent difusion pathways observed in this work suggests that a similar conclusion will be obtained even for a more realistic structure model of Mo2C with C randomly occupying half of the octahedral sites[23,24].

FIG.3 The difusion pathway of a C dopant(highlighted in yellow)in Mo2C bulk from siteβto siteγ.Energy is given in eV.Geometries of the initial,intermediate,and fnal states are illustrated in a 1×1×1 unit cell.

FIG.4(a)The Mo2C(001)surface and(b)the corresponding structure with a surface C atom lifted up as an adatom. This atom is highlighted in yellow.For both structures,top view is shown in the upper panel and side view is shown in the down panel.
B.Carbon difusion on Mo2C surfaces
Since graphene is always grown on a surface,it is thus very desirable to study carbon difusion on Mo2C surfaces as well.According to previous studies[19,25−30], the(101)surface is the most stable one.At the same time,the high-symmetry(001)surface is expected to be relevant in graphene growth[13].Therefore,both surfaces are considered in this study.To facilitate graphene growth,an Mo-terminated model is adopted for the Mo2C(001)surface.In order to study carbon difusion on these two surfaces,a 2 unit-cell slab for the(001) surface and a 1/4 unit-cell slab for the(101)surface are built.Within the surface plane,a 1×1 unit cell is large enough since the lattice parameters are larger than 6˚A.
Similar to the bulk case,we frst study the stability of C vacancy and adsorbed C atom on the surfaces.C vacancy on the(001)surface is below the topmost Mo atom layer.The(101)surface has a notable structure relaxation compared to the bulk structure,where the outmost Mo atoms shrink towards bulk by~0.40˚A and other surface atoms move upward by~0.10˚A.Such a structure relaxation smoothes the surface.If a carbon atom on the(101)surface is removed,there is no obvious further structure relaxation.As shown in Fig.2,the formation energies of C vacancy on both Mo2C(001) and(101)surfaces are positive,suggesting that forming C vacancy on these surfaces is also thermodynamically unfavorable as in the bulk case.Vacancies on the(001) surface and in the bulk have similar formation energy, which may be due to the fact that carbon locations on the(001)surface are closer to their bulk locations compared to the(101)surface.
Carbon adatom on these two surfaces has a negative formation energy,which is thermodynamically even more favorable than interstitial C doping in the bulk. Due to its Mo-terminated character,carbon adsorption on the(001)surface is 1.02 eV more favorable than on the(101)surface.C adsorption on Mo2C surfaces is more stable than C doping in the bulk.
The frst C difusion process we considered is lifting up a surface atom to get an adatom,which creates a thermodynamically unfavorable C vacancy and a favor-able C adatom simultaneously.On the Mo2C(001)surface,such a difusion process(in Fig.4 from(a)to(b)) has a 0.42 eV energy increase.The difusion barrier is 1.33 eV,which is only about half of the value in the bulk case due to the structure fexibility on the surface. On the Mo2C(101)surface,the frst surface layer contains both Mo and C atoms.If a carbon atom in the frst surface layer is pulled up(in Fig.5 from(a)to(b)), there is a 1.80 eV difusion barrier.The C adatom has a very diferent chemical environment compared to bulk C,and the fnal state is 0.44 eV higher in energy than the initial state.On both surfaces,vertical C difusion is easier than C difusion in the bulk.
Another important surface process also relevant to graphene growth is lateral C difusion on the surface. On the Mo2C(001)surface,the most stable adsorption site is a hollow site above an octahedral interstitial site (O-site)as shown in Fig.6.The neighboring metastable site is a hollow site above a tetrahedral interstitial site (T-site).From O-site to T-site,the difusion barrier is 0.91 eV and the reversed barrier is 0.40 eV.The next adsorption state is a hollow site above a C atom(C-site),which is 1.05 eV higher in energy than the O-site. The difusion barrier from T-site to C-site is 0.93 eV and the reversed barrier is 0.39 eV.In general,C difusion on(001)surface is much easier than in the bulk.
There are more metastable adsorption sites on the Mo2C(101)surface due to its lower symmetry compared to the(001)surface.The most stable adsorption structure is shown in Fig.5(c).Difusion along the[010]direction to a neighboring stable structure(Fig.5(d))has an energy barrier of 1.19 eV.Difusion from the most stable adsorption site in the[100]direction has two fnal states(Fig.7),which are 1.79 and 1.32 eV higher in energy.The difusion barrier in this direction is at least 2.30 eV,much higher than that in the[010]direction.
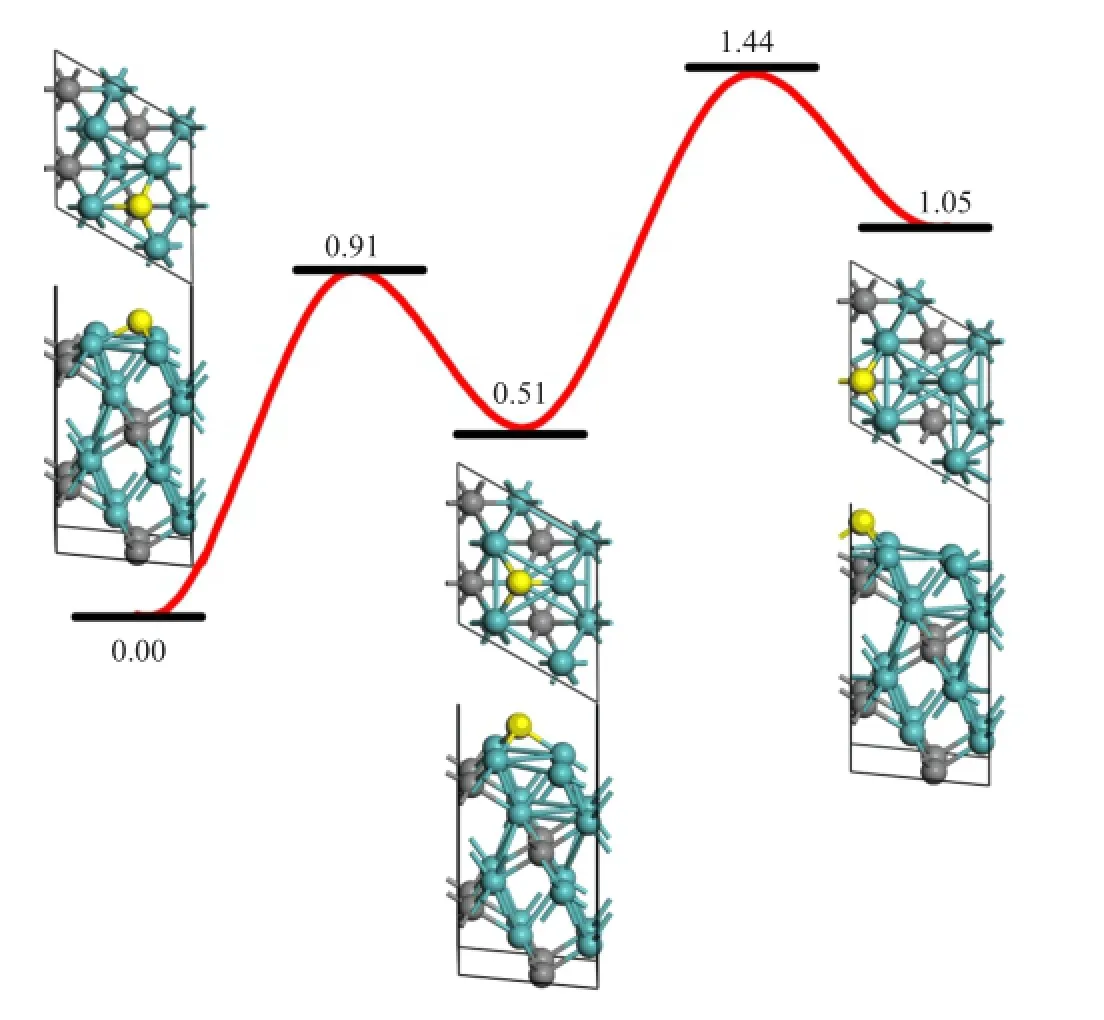
FIG.6 Difusion of C adatom(highlighted in yellow)on the (001)surface from O-site to T-site and then C-site.Relative energies compared to the most stable adsorption structure are given in eV.

FIG.7 Difusion of C adatom(highlighted in yellow)on the(101)surface in the[100]direction.Relative energies compared to the most stable adsorption structure are given in eV.
Therefore,difusion on the(101)surface is anisotropic, diferent from the(001)surface.
IV.CONCLUSION
According to our calculations,carbon difusion in Mo2C is very difcult.Although the energy barrierfor surface vacancy formation is relatively low,Mo2C surfaces are still not able to act as a sink or source for bulk vacancies,since vacancy mediated C difusion in the bulk is too difcult.The same conclusion can be applied to interstitial C dopant atoms.As a result, kinetically both C vacancy and C dopant will not be massively formed in the bulk,consisting with the high thermal stability of molybdenum carbide.Notice that the formation of C vacancy is also thermodynamically unfavorable.In the context of graphene growth,Mo2C can efectively block carbon segregation/precipitation.
Carbon difusion on Mo2C(001)surfaces typically associates an energy barrier~1 eV,which is not prohibitive at the high temperature during graphene growth.This result is consistent with the experimental observation that high quality graphene can be grown on the Mo substrate,where carbide is formed during graphene growth.On the(101)surface,C difusion is anisotropic,with the highest difusion barrier as high as 2.77 eV,which is not favorable for graphene growth. In this work,only Mo2C is studied.However,since all carbide-forming groups IVB-VIB metals show similar behaviors in the graphene growth experiment,similar conclusions are expected to be obtained for other carbides.
V.ACKNOWLEDGMENTS
This work was supported by the Ministry of Science and Technology of China(No.2014CB932700), the National Natural Science Foundation of China (No.21173202,No.21103156,and No.21222304),and the Fundamental Research Funds for the Central Universities.
[1]A.H.Castro Neto,F.Guinea,N.M.R.Peres,K.S. Novoselov,and A.K.Geim,Rev.Mod.Phys.81,109 (2009).
[2]K.S.Novoselov,V.I.Falko,L.Colombo,P.R.Gellert, M.G.Schwab,and K.Kim,Nature490,192(2012).
[3]F.Bonaccorso,A.Lombardo,T.Hasan,Z.Sun,L. Colombo,and A.C.Ferrari,Mater.Today15,564 (2012).
[4]S.Bae,H.Kim,Y.Lee,X.Xu,J.S.Park,Y.Zheng,J. Balakrishnan,T.Lei,H.R.Kim,and Y.I.Song,Nat. Nanotechnol.5,574(2010).
[5]N.C.Bartelt and K.F.McCarty,MRS Bull.37,1158 (2012).
[6]X.S.Li,W.W.Cai,J.H.An,S.Y.Kim,J.H.Nah, D.X.Yang,R.Piner,A.Velamakanni,I.H.Jung,E. Tutuc,S.K.Banerjee,L.Colombo,and R.S.Ruof, Science324,1312(2009).
[7]P.Wu,W.H.Zhang,Z.Y.Li,and J.L.Yang,Small10,2136(2014).
[8]P.Wu,H.J.Jiang,W.H.Zhang,Z.Y.Li,Z.H.Hou, and J.L.Yang,J.Am.Chem.Soc.134,6045(2012).
[9]W.H.Zhang,P.Wu,Z.Y.Li,and J.L.Yang,J.Phys. Chem.C115,22360(2011).
[10]C.P.Deck and K.Vecchio,Carbon44,267(2006).
[11]X.Li,W.Cai,L.Colombo,and R.S.Ruof,Nano Lett.9,4268(2009).
[12]L.Fu,C.H.Zhang,Y.F.Zhang,and Z.F.Liu,Acta Chim.Sin.71,308(2013).
[13]Z.Y.Zou,L.Fu,X.J.Song,Y.F.Zhang,and Z.F. Liu,Nano Lett.14,3832(2014).
[14]S.Sampath and S.F.Wayne,J.Therm.Spray Technol.3,282(1994).
[15]G.Kresse and J.Furthm¨uller,Comp.Mater.Sci.6,15 (1996).
[16]G.Kresse and J.Furthm¨uller,Phys.Rev.B54,11169 (1996).
[17]P.E.Bl¨ochl,Phys.Rev.B50,17953(1994).
[18]J.P.Perdew,K.Burke,and M.Ernzerhof,Phys.Rev. Lett.77,3865(1996).
[19]Q.Q.Luo,T.Wang,G.Walther,M.Beller,and H.J. Jiao,J.Power Sources246,548(2014).
[20]M.Methfessel and A.T.Paxton,Phys.Rev.B40,3616 (1989).
[21]G.Henkelman,B.P.Uberuaga,and H.J´onsson,J. Chem.Phys.113,9901(2000).
[22]C.Pistonesi,A.Juan,A.P.Farkas,and F.Solymosi, Surf.Sci.604,914(2010).
[23]J.Dubois,T.Epicier,C.Esnouf,G.Fantozzi,and P. Convert,Acta Metall.36,1891(1988).
[24]T.Epicier,J.Dubois,C.Esnouf,G.Fantozzi,and P. Convert,Acta Metall.36,1903(1988).
[25]T.Wang,X.W.Liu,S.G.Wang,C.F.Huo,Y.W. Li,J.G.Wang,and H.J.Jiao,J.Phys.Chem.C115, 22360(2011).
[26]J.Haines,J.Leger,C.Chateau,and J.Lowther,J. Phys.:Condens.Matter13,2447(2001).
[27]E.Rudy,S.Windisch,A.J.Stosick,and J.R.Hofman, Trans.Metall.Soc.AIME239,1247(1967).
[28]X.R.Shi,S.G.Wang,H.Wang,C.M.Deng,Z.Qin, and J.Wang,Surf.Sci.603,852(2009).
[29]X.H.Wang,H.L.Hao,M.H.Zhang,W.Li,and K. Y.Tao,J.Solid State Chem.179,538(2006).
[30]M.Nagai,A.M.Zahidul,and K.Matsuda,Appl.Catal. A313,137(2006).
10.1063/1674-0068/28/cjcp1410170is characterized by the chemical potentials of Mo and C. Considering the equilibrium with Mo2C bulk,we have
∗Author to whom correspondence should be addressed.E-mail:zyli@ustc.edu.cn
(Dated:Received on October 7,2014;Accepted on October 23,2014)
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- High Performance of Enhanced Mode Field Efect Transistor and Ultraviolet Sensor Based on ZnO Nanosheet
- Quantitative Moisture Measurement with a Cavity Ring-down Spectrometer using Telecom Diode Lasers
- Accurate Measurement of Raman Depolarization Ratio in Gaseous CO2
- Methanol Adsorption on TiO2Film Studied by Sum Frequency Generation Vibrational Spectroscopy
- Ultraviolet Source Assisted Enhancement of Attosecond Pulse
- Decay Dynamics ofN,N-Dimethylthioacetamide in S3(ππ∗)State