测定卡巴匹林钙含量的两种方法比较
2015-01-12王彦厚
董 芳,刘 敬,王彦厚*
测定卡巴匹林钙含量的两种方法比较
董 芳1,刘 敬2,王彦厚1*
目的 建立高效液相色谱法(HPLC)测定卡巴匹林钙的含量,并和容量滴定法进行对比研究。方法 色谱柱为美国热电C18柱(250 mm×4.6 mm,5 μm),流动相为磷酸-乙腈-水(2∶300∶600)溶液,进样量20 μL,检测波长为228 nm;对两种测定方法分别进行了方法学验证研究。结果 两种含量测定方法在考察浓度范围内均具有良好线性关系。结论 采用高效液相色谱法测定卡巴匹林钙的含量,该法操作简便,专属性强,结果准确。
卡巴匹林钙;高效液相色谱法;含量测定;方法比较
0 引言
卡巴匹林钙(Crbasalate calcium)为乙酰水杨酸钙与尿素的络合物,在水中水解为乙酰水杨酸而发挥解热、镇痛、抗炎和抗风湿作用[1]。该药品最先是由荷兰DSM公司研制开发(商品名:ASCAL),于1974年由荷兰卫生当局批准生产,我国于1990年批准进口卡巴匹林钙原料,目前有卡巴匹林钙散和卡巴匹林钙颗粒两种剂型[2]。卡巴匹林钙是新一代较理想的解热镇痛药。临床主要用于感冒发热、头痛、牙痛、神经痛、腰痛、肌肉痛、月经痛及风湿性关节炎等;另外,小剂量给药还可预防心脑血管疾病的发作。
卡巴匹林钙的含量测定采用的是国家药典委员会颁布的国家药品标准 WS1-(X-403)-2004Z 卡巴匹林钙中收载的容量滴定法[3]。本文参考文献[4-7]研究了采用HPLC法测定卡巴匹林钙的含量,并对两种测定方法进行了方法学验证试验和对比研究。
1 仪器与试药
岛津高效液相色谱仪(LC-10ATVP液相色谱泵,SPD-10ATVP紫外检测器,SIL-20A进样器,美国热电色谱柱C18柱(250 mm×4.6 mm,5 μm),十八烷基硅烷键合硅胶为填充剂);UV757CRT型紫外分光光度计(上海精密科学分析仪器有限公司;卡巴匹林钙(实验室自制,批号:130601、130602、130603);阿司匹林对照品(中国食品药品检定研究院,批号:100113-201104,含量:按C9H8O4计100.0%);乙腈为色谱纯,其余试剂为分析纯;水为纯化水。
2 方法与结果
2.1 容量滴定法测定含量
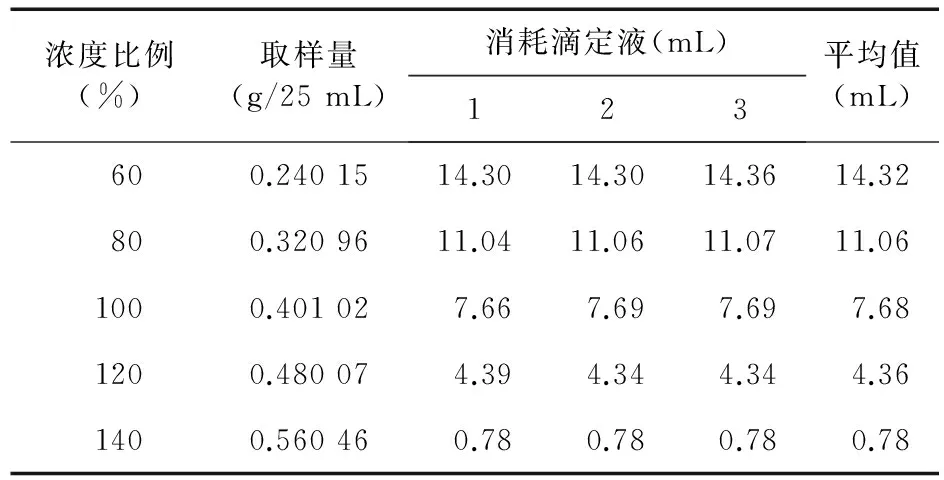
2.1.1 线性范围 精密称取本品2.4、3.2、4.0、4.8、5.6 g,分别置于250 mL量瓶中,加水溶解并稀释至刻度,作为储备液。为线性测定浓度的60%、80%、100%、120%、140%,每个储备液精密量取25 mL,置250 mL碘瓶中,精密加氢氧化钠滴定液(0.1 mol/L)25 mL,密塞,放置2 h,加酚酞指示剂3滴,用盐酸滴定液(0.1 mol/L)滴定,并将滴定结果用空白试验校正。每1 mL氢氧化钠滴定液(0.1 mol/L)相当于18.02 mg的阿司匹林(C9H8O4)。每个储备液分别取样3次,求得消耗滴定液的mL数的平均值。以样品每25 mL所含的重量为横坐标(X),以样品消耗滴定液的mL数为纵坐标(Y),线性回归,得线性回归方程Y = 42.238 X+0.272 2,r= 0.999 9(n=5)。结果见表1。
表1 容量滴定法测定含量线性范围测定结果
浓度比例(%)取样量(g/25mL)消耗滴定液(mL)123平均值(mL)600.2401514.3014.3014.3614.32800.3209611.0411.0611.0711.061000.401027.667.697.697.681200.480074.394.344.344.361400.560460.780.780.780.78
结果表明,本法测定卡巴匹林钙含量,样品取样量在0.240 15 g~0.560 46 g范围内,与消耗的氢氧化钠滴定液mL数线性关系良好。
2.1.2 重复性试验 精密量取“2.1.1”项下浓度比例为100 %的储备液25 mL,置250 mL碘瓶中,精密加氢氧化钠滴定液(0.1 mol/L)25 mL,密塞,放置2 h,加酚酞指示剂3滴,用盐酸滴定液(0.1 mol/L)滴定,并将滴定结果用空白试验校正。重复取样6次,RSD= 0.2 %。结果表明,本法测定卡巴匹林钙含量重复性良好。
2.1.3 稳定性试验 精密量取“2.1.1”项下浓度比例为100 %的储备液25 mL,置250 mL碘瓶中,室温放置,分别于0、1、2、4、8 h测定含量,含量测定结果分别为78.08%、78.08%、77.99%、77.99%、78.08%;RSD= 0.1%(n=5)。表明样品溶液在考察时间范围内有很好的稳定性。
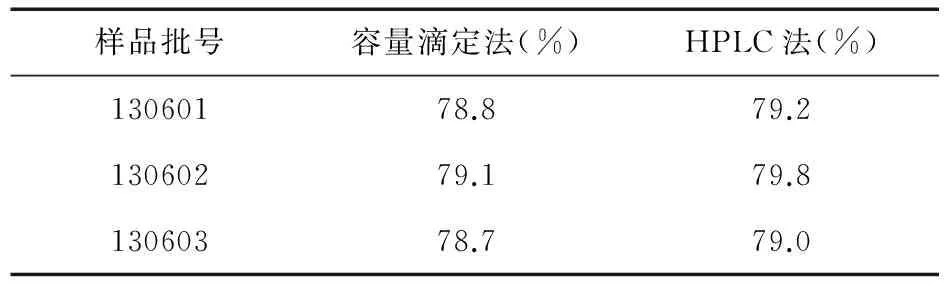
2.1.4 容量滴定法 按照国家食品药品监督管理局国家药品标准 WS1-(X-403)-2004Z 卡巴匹林钙中收载的含量测定方法(容量滴定法),对三批卡巴匹林钙样品(批号:130601、130602、130603)进行测定。取本品约 0.4 g,精密称定,置250 mL碘瓶中,加水25 mL使溶解,精密加氢氧化钠滴定液(0.1 mol/L)25 mL,密塞,放置2 h,加酚酞指示剂3滴,用盐酸滴定液(0.1 mol/L)滴定,并将滴定结果用空白试验校正。每1 mL氢氧化钠滴定液(0.1 mol/L)相当于18.02 mg的阿司匹林(C9H8O4)。结果见表2。
2.2 HPLC法
2.2.1 检测波长的确定 取“2.2.8”项下对照品溶液,以流动相为参比溶液,照紫外分光光度法在200~400 nm波长范围内扫描,结果表明本品在228 nm与277 nm处有最大吸收,选择228 nm作为检测波长。
表2 两种含量测定方法结果比较
样品批号容量滴定法(%)HPLC法(%)13060178.879.213060279.179.813060378.779.0
2.2.2 色谱条件 岛津LC-10ATVP液相色谱泵;日本岛津SPD-10ATVP紫外检测器;美国热电色谱柱C18柱(250 mm×4.6 mm,5 μm);十八烷基硅烷键合硅胶为填充剂;以磷酸-乙腈-水(2∶300∶600)为流动相;进样量20 μL,检测波长为228 nm;理论板数按阿司匹林峰计算应不低于3 000。
2.2.3 线性范围 取阿司匹林对照品适量,精密称定,用流动相制成1.0 mg/mL的溶液,作为对照品储备液。精密量取对照品储备液1.5、2.0、2.5、3.5、4.0 mL至25 mL量瓶,用流动相稀释至刻度,摇匀,分别取20 μL注入液相色谱仪,记录阿司匹林的峰面积。以样品浓度(C)为横坐标,峰面积(A)为纵坐标,线性回归,得线性回归方程为A=117 566 C+38 403,r=0.999 8(n=5)。结果表明,阿司匹林对照品在浓度0.06~0.16 mg/mL范围内具有良好的线性关系。
2.2.4 精密度试验 取线性范围项下浓度为0.1 mg/mL的溶液,精密量取20 μL,注入液相色谱仪,测定峰面积。连续进样5次,计算峰面积的RSD为0.28%(n=5)。结果表明,精密度良好。
2.2.5 重复性试验 取样品(批号:130603)采用HPLC法分别测定含量6次,平均含量为78.9%,RSD为0.19%(n=6),表明本法重现性良好。
2.2.6 回收率试验 取卡巴匹林钙样品(批号:130603)约13 mg(共9份),精密称定,分别置200 mL量瓶中,加流动相溶解,摇匀,分别精密加入阿司匹林对照品8、10、12 mg,加流动相稀释至刻度,得浓度约为样品浓度80%、100%、120%的供试品溶液,各浓度样品平行制备3份样品,摇匀,过滤,取续滤液作为供试品溶液。另取阿司匹林对照品10 mg,精密称定,置100 mL容量瓶中,流动相稀释至刻度,摇匀,作为对照品溶液。取上述两种溶液,按“2.2.2”项下的色谱条件,精密量取20 μL注入液相色谱仪,记录色谱图,按外标法以峰面积计算,即得。测得平均回收率为100.1%,RSD为0.87%(n=9)。表明本测定方法的准确度良好。见表3。
表3 卡巴匹林钙含量测定(HPLC法)回收率试验结果

样品编号加入阿司匹林对照品量(mg)测得值(mg)回收率(%)平均回收率(%)RSD(%)18.88.82100.128.68.73101.538.88.7899.8410.410.52101.1510.510.4899.8100.10.87610.410.3399.3712.412.2899.0812.112.0399.4912.812.92101.0
2.2.7 溶液稳定性试验 取线性范围项下浓度为0.1 mg/mL的溶液,于室温放置,分别于0、1、2、4、8 h取样,精密量取20 μL,注入液相色谱仪,测定峰面积。RSD为1.58 %(n=5),表明样品溶液室温放置8 h稳定。
2.2.8 含量测定(HPLC法) 取自制样品约13 mg,精密称定,置100 mL容量瓶中,流动相稀释至刻度,摇匀,作为供试品溶液,取20 μL注入液相色谱仪,记录色谱图;另取阿司匹林对照品10 mg,精密称定,置100 mL容量瓶中,流动相稀释至刻度,摇匀,作为对照品溶液;按外标法以峰面积计算。结果见表2。
3 结论与讨论
本文对容量滴定法和HPLC法测定卡巴匹林钙的含量进行了比较研究,二者方法学试验结果表明:两种测定方法均具有较好的专属性,其线性范围、稳定性、重复性等符合要求,对三批样品的含量测定结果基本一致,说明两种含量测定方法均能满足样品质量控制要求。
考虑到容量法操作繁杂,测定时可能会在判断滴定终点时产生人为误差,使测定结果有差异;目前实验室均配备了大型仪器设备,自动化程度越来越高,采用HPLC法测定含量,将样品直接用流动相溶解,操作简便、快捷,专属性强,结果准确可靠。
在采用HPLC法测定含量确定检测波长时,将阿司匹林对照品用流动相溶解后进行紫外扫描,本品在228 nm与277 nm处有最大吸收,在228 nm波长的吸光度比277 nm强,为避免噪音等不稳定因素的影响,选择228 nm作为检测波长。
[1] 蒋琪英,张玉芝,雷登武,等.乙酰水杨酸配合物的合成与应用研究现状[J].化工中间体,2007,22(8),26-30.
[2] 王素秋,徐明,觉斌温.速克痛解热镇痛作用的临床研究[J].北京医学,1998,20(3):191-192.
[3] 国家药典委员会.WS1-(X-403)-2004Z卡巴匹林钙[S].国家食品药品监督管理局国家药品标准.60:83-85.
[4] 会书钧,李怀.乙酰水杨酸锌胶囊含量的HPLC测定[J].首都医药,2001,8(11):38.
[5] 侯曙光,魏树礼.卡巴匹林钙的性质及应用[J].中国药学杂志,1996,31(2):105-107.
[6] 王晓枫,康晖,裴晓丽,等.HPLC法测定卡巴匹林镁结构中阿司匹林的含量[J].中国新药杂志,1999,8(7):463-464.
[7] 国家药典委员会.中华人民共和国药典(二部)[S].北京:中国医药科技出版社,2010:107-115.
Comparison of two methods for determination of carbasalate calcium
DONG Fang1,LIU Jing2,WANG Yan-hou1*
(1.Zibo Food and Drug Inspection & Testing Center,Zibo 255086,China; 2.Shandong Jincheng Bio-pharmaceutical Co.,Ltd.,Zibo 255130,China)
Objective To establish an HPLC method for determination of carbasalate calcium and compare with the volumetric precipitation method.Methods The Thermo C18column(250 mm×4.6 mm,5 μm)was adopted,the mobile phase was the mixture of phosphoric acid-acetonitrile-water (2∶300∶600),the injection volume was 20 μL,and the detection wavelength was 228 nm.The two methods were validated respectively.Results Two content determination methods had good linear relationship in the concentration range.Conclusion The HPLC method is simple,and with accurate and reliable results,which can be used for the determination of carbasalate calcium.
Carbasalate calcium;HPLC;Determination method;Method comparation
2015-04-22
1.淄博市食品药品检验检测中心,山东 淄博
255086;2.山东金城生物药业有限公司,山东 淄博 255130
10.14053/j.cnki.ppcr.201509021
*通信作者
