数量性状全基因组关联分析中上位效应遗传位点检测方法研究进展
2015-01-07郭家中王小龙刘海峰
郭家中,王小龙,仲 涛,刘海峰
(1 四川农业大学 动物科技学院,四川 成都611130;2 西北农林科技大学 动物科技学院,陕西 杨凌 712100)
数量性状全基因组关联分析中上位效应遗传位点检测方法研究进展
郭家中1,王小龙2,仲 涛1,刘海峰1
(1 四川农业大学 动物科技学院,四川 成都611130;2 西北农林科技大学 动物科技学院,陕西 杨凌 712100)
数量性状的表型变异受到大量效应微小的遗传位点和诸多环境因素的共同作用,但在数量性状的遗传研究领域,关于不同遗传位点之间加性效应与上位效应相对重要性的认识却存在着分歧。近年来,伴随着全基因组关联分析在人类及家养动物数量性状研究中的发展,在全基因组关联分析的框架内进行上位效应遗传位点的检测越发受到重视。文章以遗传力失踪问题为出发点,首先综述了标记-QTL连锁分析和GWAS框架下传统上位效应遗传位点的检测方法,然后对基于表型方差同质性检验和广义线性混合模型方法的上位效应统计推断以及混杂因素的处理方法进行了总结与梳理,旨在为数量性状全基因规模上位效应的相关研究提供理论参考。
数量性状;上位效应;全基因组关联分析;方差同质性
在家养动植物遗传育种中,人们关注的大多数重要经济性状属于数量性状。经典数量遗传学观点认为,数量性状的表型变异受到大量效应微小的遗传位点和诸多环境因素的共同作用。然而,长期以来对于不同遗传位点之间加性效应与上位效应(Epistasis)的相对重要性却存在着认识上的分歧[1-7]。群体内遗传方差组分的估计结果表明,大部分的遗传方差是加性效应方差[2-3,5]。另外一些学者则认为,数量性状的上位遗传效应是广泛存在的[4,6-7],但长期以来关于上位效应的相关研究却未被重视。
上位效应最早由英国遗传学家William Bateson于1909年提出,是指一个位点的等位基因效应被另一个位点的等位基因效应所掩盖的生物学现象。目前,在生物学不同分支领域中存在3种类型的上位效应,即功能上位效应(Functional epistasis)、组合上位效应(Compositional epistasis)和统计上位效应(Statistical epistasis)[8]。其中组合上位效应就是William Bateson所定义的上位效应,而统计上位效应则主要来源于Fisher关于数量性状表型值的剖分思想。广义的统计上位效应是指一个遗传位点对表型值的效应大小与遗传背景有关,而最简单的上位效应形式则是指2个遗传位点之间的相互作用[4]。在实际研究中,上位效应与基因互作效应经常被互换使用。而在数量性状QTL定位研究中,具有上位效应的数量性状遗传位点被简称为上位QTL[4](Epistatic QTL)。在过去的许多年中,尽管基于经典的QTL-标记连锁分析策略检测影响数量性状的上位QTL方法已经有所发展[9-10],但成功的家养动物数量性状上位QTL定位的研究成果并不多见[4,11]。
目前,随着全基因组关联分析研究(Genome-wide association study,GWAS)在人类和家养动物领域[12-13]不断取得的进展以及GWAS分析方法的逐步完善[14-18],在GWAS框架下进行基因互作效应的检测已引起人们越来越多的兴趣。虽然一些常用的GWAS统计分析模型和软件(PLINK[14]、GenABEL[15])也考虑了上位效应的检测,但在应用中主要还是针对加性效应的检测。最近,户国等[19]对上位效应的概念起源及其对家养动物重要经济性状的遗传影响进行了总结;郭家中等[20]针对家养动物数量性状的加性效应遗传位点的GWAS单标记分析策略进行了论述;栾奕昭等[21]讨论了如何采用数据挖掘算法分析患病性状基因的互作。然而,关于利用统计建模和推断对上位QTL进行定位的研究尚缺乏详细的探讨。本研究以失踪遗传力问题作为出发点,首先综述了标记-QTL连锁分析和GWAS框架下传统上位效应遗传位点的检测方法,然后对基于表型方差同质性检验和广义线性混合模型方法的上位效应统计推断以及混杂因素的处理方法进行了总结与梳理,旨在为数量性状全基因规模上位效应的相关研究提供理论参考。
1 遗传力失踪问题
在过去的几年中,针对人类数量性状和复杂疾病的GWAS取得了丰硕的成果,鉴定出大量新的遗传位点[12]。尽管如此,这些位点共同解释的遗传方差仅占到遗传力的一小部分,剩余的大部分遗传力无法解释,从而形成“遗传力失踪”(Missing heritability)的遗传学问题[22]。受此影响,GWAS的理论假设和依据遭到一些研究者的质疑[23]。
针对遗传力失踪问题,Manolio等[24]进行了详细讨论,并提出遗传力失踪的主要原因包括:(1)大多数遗传位点对数量性状的表型效应太小,由于统计分析功效的限制,此类位点很难检测,例如人类的身高性状[25];(2)数量性状的表型变异也可能是由基因组结构的变化所引起的,如拷贝数变异(Copy number variation)或结构变异(Structural variation);(3)数量性状的表型变异也可能受到稀有变异(Rare variants),即少数等位基因频率小于0.5%的遗传变异的影响,而由于理论假设的限制,GWAS方法无法检测这类变异[26];(4)数量性状的表型变异还可能来源于多个遗传位点之间的相互作用,即上位效应。而关于上位效应的重要性则被Paré等[27]、Bloom等[28]的报道进一步证实。另外,Zuk等[29]提出基因加性效应方差会因基因互作效应而高估,从而造成遗传力被低估,该观点尝试从遗传力定义中分母的角度解释遗传力失踪的可能原因。
2 上位效应位点检测的直接方法
2.1 标记-QTL连锁分析中的上位效应检测
相对于数量性状加性效应的分析,由于统计学上位效应和生物学上位效应之间的差异,上位效应遗传分析和理论解释并不统一[8,30]。在数量性状的遗传分析中,大家主要讨论2个位点间的互作这一最简单的上位效应形式,也就是将2个位点的总体基因型值与2个位点的边缘加性效应之和的离差定义为统计上位效应[8](图1),如图1-A显示了双位点加性效应模式;而图1-B则展示了双位点显性上位效应作用:对于某一数量性状只有当第1个位点的显性等位基因不存在时,第2个位点不同基因型控制的表型均值才具有统计上的显著性差异。而包含了互作效应项的线性模型分析方法通常也被称为上位效应分析的直接方法,对于双位点上位效应的检测可以采用双因素方差分析(Two-way ANOVA)比较以下2个模型[31]或直接根据模型(2)进行互作效应的估计和推断:
yij=μ+αi+βj+eij。
(1)
yijk=μ+αi+βj+γij+eij。
(2)
式中:yij、yijk均为数量性状表型值;μ为对环境效应进行校正后的总体均值,αi为A位点的基因型效应,βj为B位点的基因型效应,eij为服从标准正态分布的残差,γij为A和B位点的基因型互作效应。
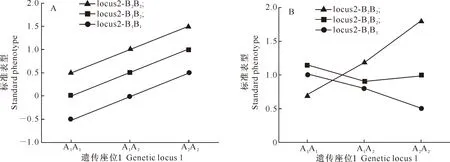
图1 数量性状双位点加性效应和显性上位效应示意图
尽管模型(2)是双位点上位效应分析的常用模型,但在全基因组规模的研究中,还要面临多重检验的问题。考虑到上位效应的性质,如果使用传统的Bonferroni方法进行多重检验的校正则过于保守,可能会遗漏一些真实的位点。针对标记-QTL连锁分析中上位效应的统计检验,Carlborg等[32]提出了上位QTL分析的随机化检验方法,即基于分析的数据直接构造经验分布。采用上述方法,Carlborg等[11]以赤色原鸡与白来航鸡杂交所获得的F2个体作为资源群体,首次检测到6个控制鸡生长性状的上位效应遗传位点;随后,Carlborg等[33]、Besnier等[34]和Pattersson等[35]围绕影响不同周龄鸡体质量的上位QTL开展了系列研究,其中有5个上位QTL在F8世代仍然可以被检测到。
上述多世代样本的遗传分析说明,上位效应是可以稳定遗传的,这也是迄今为止畜禽重要经济性状上位效应的定位中最成功的研究成果。众所周知,动物的毛色性状也被认为受上位遗传效应的控制,但该性状属于典型的质量性状。户国等[19]对控制动物毛色性状不同座位的相互关系进行了讨论。总体来说,基于传统标记-QTL分析的上位QTL研究报道并不多见。究其原因,主要是因为影响数量性状表型的统计上位遗传效应非常小而难以检测,尤其以低密度的微卫星标记作为分析基础时更不容易检测。但随着基因组测序技术的发展,高密度的SNP分型芯片的成功研制为数量性状QTL定位提供了新的机遇。
2.2 GWAS框架下上位效应位点的检测
与传统的标记-QTL连锁分析只能利用家系内的信息相比,GWAS理论上能够充分利用群体内包含的所有重组信息,因此该方法有着更高的统计功效[36]。自2005年被成功用于定位与影响人类年龄相关的视网膜黄斑的遗传变异位点[37]以来,GWAS就迅速地在人类复杂疾病的遗传研究中得到应用并取得了巨大成功[12]。此后,随着各物种高密度SNP芯片的陆续开发,该方法又被应用到家养动植物和模式生物数量性状的遗传研究中[12,38-39]。
在基于单标记回归分析策略的数量性状GWAS方法或模型[13-17]发展成熟后,尤其受到“遗传力失踪”问题的影响,如何鉴定具有上位效应的遗传位点就成为复杂疾病或数量性状GWAS方法领域发展的重点[40]。事实上,Purcell等[14]开发的PLINK软件中已经包含了双位点互作的上位效应分析命令。然而,大规模上位效应具体分析中的最大障碍却是计算量问题。简单来说,两两互作的二维全基因组分析的计算时间大约是一维单位点加性效应模型所需时间的平方倍。目前,各物种高密度SNP芯片至少包含几万个位点,例如猪的Illumina Porcine SNP60K Beadchip、牛的Illumina Bovine SNP50 Beadchip等商业芯片。如果采用上述密度的SNP芯片,全基因组上位效应分析将需要较长的计算时间。例如,Cordell[40]在单节点计算机群上应用PLINK软件的“——fast-epistasis”命令,最终运行14 d才能完成约90 000个SNPs之间两两互作效应的分析。而Wang等[41]推算若在单核计算机上采用EPISNP[42]软件对500 000个SNPs进行两两互作效应的分析,则需要1.2年才能完成。类似地,理论上3个遗传位点之间互作效应分析所需要的计算时间则是SNPs位点数目的立方倍。因此,在基于高密度SNP芯片数据的实际研究中,若采用上位效应直接检测方法分析3个以上遗传位点的互作效应是不现实的。
为了解决计算量问题,Schüpbach等[43]在PLINK软件上位效应分析模块的基础上发展了FastEpistasis软件。而生物信息学研究人员采用不同的机器学习(Machine Learning)算法,又陆续开发了SNPHarvester[44]、TEAM[45]、BOOST[46]、SNPRuler[47]、Screen and Clean[48]等程序或方法。相对而言,TEAM在单个位点具有主效应的数据分析中统计功效最高;而BOOST则对单位点无主效应的数据统计功效最高。另外,前4种方法是单阶段分析方法;而Screen and Clean模型则采用两步法,其中第二阶段只是针对第一阶段分析检测到的显著性SNPs进行全基因组上位效应分析,通过正向选择策略总体减少了计算时间,但相对于前4种方法,该方法假阳性率最高[41]。另外,有观点认为[8,49],以单标记显著性检验为基础的两步法并不是非常合理的,因为上位效应可能发生在那些效应非常小的遗传位点之间,甚至发生在那些根本就没有主效应的位点之间。最近,Hemani等[50]以经典的数量性状遗传分析模型为基础,利用GPU硬件技术发展了双位点穷尽式上位效应遗传分析软件epiGPU,理论上利用GeForce GTX 580图形处理器使用 epiGPU将较基于CPU技术的计算机快90倍。Hemani等[51]利用此软件,采用双阶段分析策略,在846个人类群体样本中以7 339个基因表达丰度作为表型,进行全基因组规模双位点上位效应关联分析,共检测到500多个双位点上位效应,显示了该程序在计算效率上的优势。
3 基于方差同质性检验的上位效应分析
3.1 方差同质性检验的非参数方法
如上所述,尽管包括FastEpistasis 和epiGPU在内的一些以模型(2)为理论基础的高效率计算软件或程序[42-47,49]已经陆续被开发出来,但在GWAS中尚未得到广泛应用。而从统计推断的角度,理论上多个(≥2)遗传位点之间的互作效应会导致单个位点表型方差的异质性,即不同基因型组间的表型方差具有显著性差异[27,52]。更重要的是,从计算效率的角度,针对单个位点开展表型同质性检验是上位效应遗传位点检测的捷径[52-53]。因此,在统计遗传学领域,近期关于上位效应的检测方法主要围绕方差同质性检验而展开。
传统的方差同质性检验主要有两种方法:一是围绕样本观察值与组内均值的离差构造的Bartlett检验;二是以样本观察值与组内中位数的离差为基础而构造的Levene检验(即Brown-Forsythe检验)。与中位数相比较,许多情况下均值更容易受到样本中极端值的影响,所以Levene检验比Bartlett检验更稳健。总的来说,这两种方法均属于非参数检验,且构造的检验统计量均服从F分布。当分母自由度非常大时,F分布近似等价于分子自由度的χ2分布。Levene检验可采用下面的公式表示:
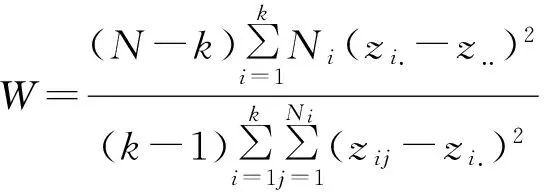
(3)
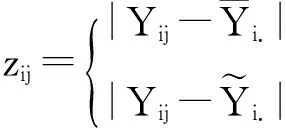
在基于群体水平设计的数量性状GWAS中,Paré等[27]、Struchalin等[52]最早同时提出将方差同质性统计检验方法作为上位效应遗传位点的分析策略。其中Struchalin等[52]从理论上对基因互作效应估计的直接法和方差异质性检验的间接法进行了比较,表明直接检验的统计功效高于间接方法,且方差同质性检验的功效仍受到单个位点主效应的影响;对于符合正态分布的经典数量性状,Bartlett检验、通过秩转换后的Bartlett检验(Bartlett test with prior rank transformation to normality)以及Levene检验3种方法中,Bartlett检验拥有最高的统计功效,而当性状偏离正态分布时,Levene检验的统计功效更高。Struchalin等[54]开发了R环境下的GWAS方差异质性分析软件包VariABEL,其中包含了Bartlett检验和Levene检验2种方法。而Paré等[27]基于方差同质性检验,发现了影响妇女炎症标记含量的基因互作位点,用实例说明了方差异质性分析是上位效应检测的一种有效方法。尽管如此,Shen等[55]通过模拟发现,在针对不平衡数据的GWAS中,由于方差的分布偏离正态性、方差理论分布的下限为零和方差与均值的相关性等,基于Bartlett检验、Levene检验或平方Z-score检验(Squared Z-score)的表型方差异质性统计推断将存在非常高的假阳性率。另外,Shen等[56]还发现,在检测控制表型方差的遗传位点研究中,表型数据的刻度水平对统计功效有很大影响,应谨慎使用任何形式的转换方法对原始数据进行转换。
3.2 群体混杂因素的考虑
众所周知,群体水平的全基因组关联研究必须考虑群体分层及多个亲缘关系较近样本个体对统计检验的干扰。显然,上述因素也同样会影响群体水平的遗传方差同质性检验。当混杂因素的干扰较低时,考虑到方差同质性检验服从χ2分布,仍可采用基因组控制的校正方法[57]。但是在家养动植物数量性状的遗传分析中,面对亲缘关系造成的高强度混杂,仅使用基因组控制方法进行关联结果的校正则不够合理。针对此问题, Rönnegård等[58]和Shen等[59]根据层次似然理论(Hierarchical likelihood,h-likelihood),先后发展出基于层次广义线性模型(Hierarchical Generalized Linear Models)的R软件包hglm和广义岭回归方法(Generalized Ridge Regression)R软件包bigRR。上述方法的主要原理是通过在残差部分中拟合加性随机效应从而达到对群体混杂的校正。因此,在未来的全基因组方差异质性研究中,上述方法很有可能被广泛采用。
在传统的GWAS框架中,与群体水平的研究不同,基于家系的关联或连锁分析则从试验设计上排除了混杂因素对统计推断的干扰[60]。因此,基于家系设计的数量性状同类研究在实践中经常出现。最近,Rönnegård等[61]发展了适合于F2或回交设计连锁分析的方差异质性QTL检测方法,该方法基于双层广义线性模型理论(Double generalized linear model,dglm),通过分别估计遗传协变量对表型的均值和方差的效应,可同时检测控制表型均值的普通QTL及影响表型方差的vQTL(Variance-controlling QTL,vQTL)。总体而言,与Bartlett检验、Levene检验不同,Rönnegård等[61]的方法属于参数检验,尤其对于经典的数量性状统计功效更高。
3.3 表型方差异质性与上位效应的关系
基于方差同质性检验上位效应遗传位点的检测策略近期得到了关注及发展,但值得注意的是,方差异质性的存在并不一定仅仅是指发生在2个位点间的上位效应,同时也表明可能有一个复杂的调控网络存在[53]。另外,数量性状表型方差异质性也可能是由遗传因素与环境协变量之间的相互作用造成的;近年来,在动物育种研究领域,由遗传因素与环境间的互作造成的表型方差异质性也越来越引起大家的兴趣[62-64]。简言之,上位效应与方差异质性之间是充分非必要的关系[52-53]。尽管如此,由于将二维或高维(多个位点间的互作效应)的全基因组分析简化为一维分析,以基因型分组为基础的方差同质性检验是一种高效的上位效应遗传位点研究策略。
4 结 语
长期以来,在数量遗传学和群体遗传学以及相关领域中,关于上位效应的重要性一直存在争论[1-7]。3种上位效应的定义以及内涵的差异[8]造成了上位效应理解的困难与混乱,最终导致上位效应的遗传研究被长期忽视。自2008年以来,受到“遗传力失踪”[22]这一科学问题的影响,在全基因组关联分析框架下数量性状上位效应的遗传研究逐渐受到了重视,各种高效率上位效应分析方法和软件相继被提出,尤其是基于表型方差同质性的检验分析策略被视作全基因组规模上位效应分析的一条捷径。与此同时,一些研究小组[51,65]以表达谱芯片技术测定的基因表达丰度作为表型值并结合高密度SNP芯片,成功鉴定出多个互作效应位点;另外,也有学者将数量性状表型值的变异系数(Coefficient of variation,CV)作为因变量,鉴定了控制相关性状表型变异系数变化的遗传位点[66-67];而 Jimenez-Gomez等[68]同时采用上述2种策略鉴定出多个影响拟南芥相关代谢性状的遗传位点。上述研究策略为数量性状的上位效应研究开辟了新的思路。随着SNP分型和表达谱芯片成本的整体下降,这一类型的上位效应研究也许会越来越多。
总体来说,伴随着统计模型的发展、计算机计算效率的不断提高以及高密度SNP芯片和高通量测序技术的不断发展,家养动物数量性状全基因组规模的上位效应研究将进入新的阶段,所获得的结果将会促进对数量性状表型变异遗传机制的更深入理解。
[1] Wright S.Evolution in mendelian populations [J].Genetics,1931,16(2):97-159.
[2] Falconer D S,Mackay T F C.Introduction to quantitative genetics [M].4th Edition.England:Longman,1996:129-140.
[3] Lynch M,Walsh B.Genetics and analysis of quantitative traits [M].Sunderland,MA:Sinauer Associates,1998:65-71.
[4] Carlborg O,Haley C S.Epistasis:Too often neglected in complex trait studies? [J].Nat Rev Genet,2004,5(8):618-625.
[5] Hill W G,Goddard M E,Visscher P M.Data and theory point to mainly additive genetic variance for complex traits [J].PLoS Genet,2008,4(2):e1000008.
[6] Breen M S,Kemena C,Vlasov P K,et al.Epistasis as the primary factor in molecular evolution [J].Nature,2012,490(7421):535-538.
[7] Hemani G,Knott S,Haley C.An evolutionary perspective on epistasis and the missing heritability [J].PLoS Genet,2013,9(2):e1003295.
[8] Phillips P C.Epistasis:The essential role of gene interactions in the structure and evolution of genetic systems [J].Nat Rev Genet, 2008, 9(11):855-867.
[9] Haley C S,Knot S A.A simple regression method for mapping quantitative trait loci in line crosses using flanking markers [J].Heredity,1992,69(4):315-324.
[10] Kao C H,Zeng Z B,Teasdale R D.Multiple interval mapping for quantitative trait loci [J].Genetics,1999,152(3):1203-1216.
[11] Carlborg O,Jacobsson L,Ahgren P,et al.Epistasis and the release of genetic variation during long-term selection [J].Nat Genet,2006,38(4):418-420.
[12] Visscher P M,Brown M A,McCarthy M I,et al.Five years of GWAS discovery [J].Am J Hum Genet,2012,90(1):7-24.
[13] Andersson L S,Larhammar M,Memic F,et al.Mutations in DMRT3 affect locomotion in horses and spinal circuit function in mice [J].Nature,2012,488(7413):642-646.
[14] Purcell S,Neale B,Todd-Brown K,et al.PLINK:A tool set for whole-genome association and population-based linkage analyses [J].Am J Hum Genet,2007,81(3):559-575.
[15] Aulchenko Y S,Ripke S,Isaacs A,et al.GenABEL:An R library for genome-wide association analysis [J].Bioinformatics,2007,23(10):1294-1296.
[16] Aulchenko Y S,de Konning D J,Haley C.Genome-wide rapid association using mixed model and regression:A fast and simple method for genome-wide pedigree-based quantitative trait loci association analysis [J].Genetics,2007,177(1):577-585.
[17] Kang H M,Zaitlen N A,Wade C M,et al.Efficient control for population structure in model organism association mapping [J].Genetics,2008,178(3):1709-1723.
[18] Zhang Z W,Ersoz E,Lai C Q,et al.Mixed linear model approach adapted for genome-wide association studies [J].Nat Genet,2010,42(4):355-360.
[19] 户 国,王守志,李 辉.上位性及其对家养动物复杂经济性状形成的遗传学影响 [J].东北农业大学学报,2012,43(3):117-122.
Hu G,Wang S Z,Li H.Epistasis and its genetic influence on phenotype shaping of complex economic traits in domestic animals [J].Journal of Northeast Agricultural University,2012, 43(3):117-122. (in Chinese)
[20] 郭家中,王小龙,刘小林.家养动物数量性状全基因组关联研究进展:单标记回归分析策略 [J].家畜生态学报,2013,34(8):1-5.
Guo J Z,Wang X L,Liu X L.Advancement of genome-wide association study for quantitative traits in farm animals:Strategy of single-marker regression analysis [J].Acta Ecologiae Animalis Domastici,2013,34(8):1-5.(in Chinese)
[21] 栾奕昭,左晓宇,刘 轲,等.基于单核苷酸多态性的基因互作分析方法学进展 [J].遗传,2013,35(12):1331-1339.
Luan Y Z,Zuo X Y,Liu K,et al.Advances in development of gene-gene interaction analysis methods based on SNP data: A review [J].Hereditas,2013,35(12):1331-1339.(in Chinese)
[22] Mahar B.The case of the missing heritability [J].Nature,2008,456(7218):18-21.
[23] McClellan J,King M C.Genetic heterogeneity in human diseases [J].Cell,2010,141:210-217.
[24] Manolio T A,Collins F,Cox N J,et al.Finding the missing heritability of complex diseases [J].Nature,2009,461(7265):747-753.
[25] Yang J,Benyamin B,Mcevoy B P,et al.Common SNPs explain a large proportion of the heritability of human height [J].Nat Genet,2010,42(7):18-21.
[26] Bansal V,Libiger O,Torkamani A,et al.Statistical analysis strategies for association studies involving rare variants [J].Nat Rev Genet,2010,11(11):773-785.
[27] Paré G,Cook N R,Ridker P M.On the use of variance per genotype as a tool to identify quantitative trait interaction effects:A report from the Women’s Genome Health Study [J].PLoS Genet,2010,6(6):e1000981.
[28] Bloom J S,Ehrenreich I M,Loo W T,et al.Finding the sourc-es of missing heritability in a yeast cross [J].Nature,2013,494(7436):234-237.
[29] Zuk O,Hechter E,Sunyaev S R,et al.The mystery of missing heritability:Genetic interaction create phantom heritability [J].Proc Natl Acad Sci USA,2011,109(4):1193-1198.
[30] Alvarez-Castro J M,Carlborg O.A unified model for functional and statistical epistasis and its application in quantitative trait loci analysis [J].Genetics,2007,176(2):1151-1167.
[31] Shen X.Novel statistical methods in quantitative genetics [D].Uppsala (Sweden):Uppsala University,2012.
[32] Carlborg O,Andersson L.Use of randomization testing to detect multiple epistatic QTLs [J].Genet Res,2002,79(2):175-184.
[33] Carlborg O,Kerje S,Schütz K,et al.A global search reveals epistatic interaction between QTL for early growth in the chicken [J].Genome Res,2003,13(3):413-421.
[34] Besnier F,Wahlberg P,Ronnergard L,et al.Fine mapping and replication of QTL in outbred chicken advanced intercross lines [J].Genet Sel Evol,2011,43:3.
[35] Pettersson M,Besnier F,Siegel P,et al.Replication and explorations of higher-order epistasis using a large advanced intercross line pedigree [J].PLoS Genet,2011,7(7):e1002180.
[36] Hirschhorn J N, Daly M J.Genome-wide association studies for common diseases and complex traits [J].Nat Rev Genet,2005,6(2):95-108.
[37] Klein R J,Zeiss C,Chew E Y,et al.Complement factor H polymorphism in age-related macular degeneration [J].Science,2005,308(5720):385-389.
[38] Atwell S,Huang Y S,Vilhjalmsson B J,et al.Genome-wide association study of 107 phenotypes inArabidopsisthalianainbred lines [J].Nature,2010,465(7298):627-631.
[39] Zhao K,Tung C W,Eizenga G C,et al.Genome-wide association mapping reveals a rich genetic architecture of complex traits inOryzasativa[J].Nat Commun,2011,2:467.
[40] Cordell H J.Detecting gene-gene interactions that underlie human diseases [J].Nat Rev Genet,2009,10(6):392-404.
[41] Wang Y,Liu G,Feng M,et al.An empirical comparison of several recent epistatic interaction detection methods [J].Bioinformatics,2011,27(21):2936-2943.
[42] Ma L,Runesha H B,Dvorkin D,et al.Parallel and serial computing tools for testing single-locus and epistatic SNP effects of quantitative traits in genome-wide association studies [J].BMC Bioinformatics,2008,9:315.
[43] Schüpbach T,Xenarios I,Bergmann S,et al.FastEpistasis:A high performance computing solution for quantitative trait epistasis [J].Bioinformatics,2010,26(11):1468-1469.
[44] Yang C,He Z,Wan X,et al.SNPHarvester:A filtering-based approach for detecting epistatic interactions in genome-wide association studies [J].Bioinformatics,2009,25(4):504-511.
[45] Zhang X,Huang S,Zou F,et al.TEAM:Efficient two-locus epistasis tests in human genome-wide association study [J].Bioinformatics,2010,26(12):217-227.
[46] Wan X,Yang C,Yang Q,et al.BOOST:A fast approach to detecting gene-gene interactions in genome-wide case-control studies [J].Am J Hum Genet,2010,87(3):325-340.
[47] Wan X,Yang C,Yang Q,et al.Predictive rule inference for epistatic interaction detection in genome-wide association studies [J].Bioinformatics,2010,26(1):30-37.
[48] Wu J,Devlin B,Ringquist S,et al.Screen and clean:A tool for identifying interactions in genome-wide association studies [J].Genet Epidemiol,2010,34(3):275-285.
[49] Xu S,Jia Z.Genome wide analysis of epistatic effects for quantitative traits in barley [J].Genetics,2007,175(4):1955-1963.
[50] Hemani G,Theocharidis A,Wei W,et al.EpiGPU:Exhaustive pairwise epistasis scans parallelized on consumer level graphics cards [J].Bioinformatics,2011,27(11):1462-1465.
[51] Hemani G,Shakhbazov K,Westra H J,et al.Detection and replication of epistasis influencing transcription in humans [J].Nature,2014,508(7495):249-253.
[52] Struchalin M V,Dehghan A,Witteman J C,et al.Variance he-terogeneity analysis for detection of potentially interacting genetic loci:Method and its limitations [J].BMC Genet,2010,13(11):92.
[53] Rönnegård L,Valdar W.Recent developments in statistical me-thods for detecting genetic loci affecting phenotypic variability [J].BMC Genet,2012,13:63.
[54] Struchalin M V,Amin N,Eilers P H,et al.An R package “VariABEL” for genome-wide searching of potentially interacting loci by testing genotypic variance heterogeneity [J].BMC Genet,2012,13:4.
[55] Shen X,Carlborg Ö.Beware of risk for increased false positive rates in genome-wide association studies for phenotypic variability [J].Front Genet,2013,4:93.
[56] Shen X,Rönnegård L.Issues with data transformation in genome-wide association studies for phenotypic variability [J].F1000Res,2013,2:200.
[57] Shen X,Pettersson M,Rönnegård L,et al.Inheritance beyond plain heritability:Variance-controlling genes inArabidopsisthaliana[J].PLoS Genet,2012,8(8):e1002839.
[58] Rönnegård L,Xia S,Moudud A.hglm:A package for fitting hierarchical generalized linear models [J].The R Journal,2010,2:20-28.
[59] Shen X,Alam M,Fikse F,et al.A novel generalized ridge regression method for quantitative genetics [J].Genetics,2013,193(4):1255-1268.
[60] Laird N M,Lange C.Family-based designs in the age of large-scale gene-association studies [J].Nat Rev Genet,2006,7(5):385-394.
[61] Rönnegård L,Valdar W.Detecting major genetic loci controlling phenotypic variability in experimental crosses [J].Genetics,2011,188(2):435-447.
[62] Rönnegård L,Felleki M,Fikse W F,et al.Variance component and breeding value estimation for genetic heterogeneity of residual variance in Swedish Holstein dairy cattle [J].J Dairy Sci,2013,96(4):2627-2636.
[63] Sonesson A K,Odegård J,Rönnegård L.Genetic heterogeneity of within-family variance of body weight in Atlantic salmon (Salmosalar) [J].Genet Sel Evol,2013,45:41.
[64] Mulder H A,Rönnegård L,Fikse W F.Estimation of genetic variance for macro- and micro-environmental sensitivity using double hierarchical generalized linear models [J].Genet Sel Evol,2013,45:23.
[65] Wang G,Yang E,Brinkmeyer-Langford C L,et al.Additive,epistatic,and environmental effects through the lens of expression variability QTL in a Twin Cohort [J].Genetics,2014,196(2):413-425.
[66] Le Rouzic A.Estimating directional epistasis [J].Front Genet,2014,5:198.
[67] Joseph B,Corwin J A,Kliebenstein D J.Genetic variation in the nuclear and organellar genomes modulates stochastic variation in the metabolome,growth,and defense [J].PLoS Genet,2015,11(1):e1004779.
[68] Jimenez-Gomez J M,Corwin J A,Joseph B,et al.Genomic analysis of QTLs and genes altering natural variation in stochastic noise [J].PLoS Genet,2011,7(9):e1002295.
Advances in statistical approaches for detection of epistatic genetic loci for genome-wide association study of quantitative traits
GUO Jia-zhong1,WANG Xiao-long2,ZHONG Tao1,LIU Hai-feng1
(1CollegeofAnimalScienceandTechnology,SichuanAgriculturalUniversity,Chengdu,Sichuan611130,China; 2CollegeofAnimalScienceandTechnology,NorthwestA&FUniversity,Yangling,Shaanxi712100,China)
The controversy on the relative importance between additive effects and epistasis in genetics research of quantitative traits has existed for a long time,although phenotypic variations for such traits are generally believed to be governed by polygenic genes and many environmental factors.With the recent advancement of genome-wide association study of quantitative traits in humans and domesticated animals,the identification of epistasis or genetic interaction in the framework of genome-wide association study attracted increasing attention.With the issue of missing heritability as the starting point,we firstly reviewed and clarified the conventional approaches for detection of epistatic genetic loci in marker-QTL linkage analyses and genome-wide association studies.Then,we summarized the methods of homogeneity tests,the generalized linear mixed model for both phenotypic variance of quantitative traits,and adjustment of confounding factors.The paper would provide reference for genome-wide epistatic QTL studies on quantitative traits.
quantitative traits;epistasis;genome-wide association study;variance homogeneity
时间:2015-09-09 15:41
10.13207/j.cnki.jnwafu.2015.10.002
2014-03-21
四川省教育厅重点项目(13ZA0264);教育部留学回国人员科研启动基金项目
郭家中(1982-),男,江苏徐州人,讲师,博士,主要从事动物遗传育种研究。E-mail:jiazhongguo2003@gmail.com
刘海峰(1974-),男,四川巴中人,副教授,博士,主要从事动物遗传育种研究。E-mail:liuhf0-1@126.com
S813.1
A
1671-9387(2015)10-0007-07
网络出版地址:http://www.cnki.net/kcms/detail/61.1390.S.20150909.1541.004.html
